









 您已经拒绝加入团体
您已经拒绝加入团体

 2021-10-18
2021-10-18
 17242
17242
 0
0
【摘要】 XAFS、XANES、EXAFS
从上世纪七十年代开始,基于同步辐射的X射线吸收谱学技术逐渐发展成熟起来,并迅速成为研究凝聚态物质结构的新工具,尤其是在多相催化领域有重要应用。其中,X射线精细结构吸收谱(XAFS),包括近边吸收结构(XANES)和拓展边吸收结构(EXAFS),已经成为材料科学和催化领域重要的表征工具。XAFS技术对中心吸收原子的局域结构和化学环境十分敏感,因而能够在原子尺度上表征某原子邻近几个配位壳层的结构信息,如配位原子的种类、配位数、无序度、与中心原子的距离、氧化态等。除了同步辐射技术外,荧光XAFS也可以用于研究百万分之几低浓度的样品和几个原子层厚度的薄膜样品,磁XAFS用于研究材料的电子自旋状态,高温和高压的原位XAFS研究材料的相变过程,空间分辨XAFS研究材料的微区结构,时间分辨XAFS研究反应的动力学等。
1.XAFS的发展
瑞典Siegbahn和Stenstrom在1916首次发展出真空X射线光谱,见图1a。1920年,Fricke首次在实验中观测到选定原子的精细结构吸收K边[15],Hertz在同年观测到了选定原子的L边。1931年,Hanawalt通过XAS精细结构谱观测到了样品的化学和物理态。将XAFS光谱置于摄影底片,见图1b。他用XAFS表征样品在固相和液相的不同精细结构,证明了物质在分子形式的升华过程,诸如(4Assolid→ (As4)gas)或AsCl3,见图1c。接着,他观测发现,Zn、Hg、Xe和Kr的单原子蒸汽,见图1d,没有二级结构。在1931-1932,Kronig基于长城有序系统第一次尝试从理论上解释XAS谱精细结构,将晶体中的XAFS振荡归结为态密度的影响。然而由于假设的错误,这个理论是不完整的,它无法解释在气体、液体、溶液和非晶形固体中观测到的EXAFS信号。直到1932,Kronig受Hanawalt实验启发,基于短程有序提出了一套解释双原子分子精细结构谱的新理论。该理论将XAFS的振荡结构归因于周围原子对光电子跃迁矩阵元的影响。尽管该理论无法提供样品吸收原子局域结构的定量信息,它仍揭示了XAFS的基本概念。一直从上世纪三十年代到六十年代,XAFS仍只是一个光谱学的一个兴趣,并没有真正成为一项重要的表征技术。在大多数研究中,讨论仅限于列出给定材料精细结构的最大值和最小值,并将这些值与不同理论的预测值相比较,无法提取定量信息,只能获得定量结论。
在上世纪六十年代,XAFS仪器取得重大突破。商业衍射仪得以改进,因而在仍用穿欧婷X射线管作光源,也可以获得更高质量的吸收谱。借助改进的仪器,Van Nordstrand首次应用XANES于催化研究。1971年Sayers、Stern和Lytle做出了开创性工作,他们对k空间的谱图做傅里叶变换,成功地把不同原子配位壳层的信号分离了开来。他们能够从EXAFS谱中获取到晶相和非晶相Ge的定量信息。此项工作是EXAFS中的开创性工作,在70年代,他们基于Green函数和松饼模型散射势进行了进一步推演。从1970开始,EXAFS和XANES逐渐成为了探测未知系统结构和电子构型的重要工具,这主要归功于同步辐射技术的应用。同步辐射具有高亮度、大范围连续波长可调、很好的偏振性和很小的发射角的优越特点。1974年Stern等首次用同步辐射光源采集XAFS谱。随着原位XAFS技术的发展,EXAFS和XANES成为研究催化领域的关键技术。
图1.XAFS示意图
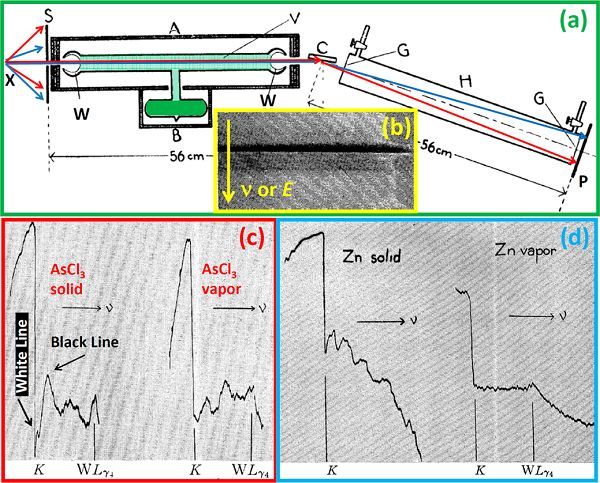
(a)XAFS原理图;(b)XAFS电镜图;(c)固态和气态AsCl3谱图;(d)固态和气态Zn谱图。
2.XAFS的原理
2.1 单散射近似
XAFS的产生原理为:物质对X射线的吸收是一个光电离过程,原子吸收X光子后,内层电子被激发出来,形成向外出射的光电子波.此波在向外传播过程中,受到邻近原子的作用而被散射,散射波与出射波的相互干涉改变了原子的电子终态波函数,导致在高能侧原子对X射线的吸收出现振荡现象,见图2。为了从吸收截面中分离出结构信息,定义EXAFS光谱函数χ为的μ(E)震荡部分,可表示为:
χ(k) = [μ(k) - μ0(k)]/μ0(k) (1)
k为光电子波矢量,可表示为:
k= = (2)
其中,m和Ef是光电子的质量和动能,为入射光子的能量,Eb是光电子的结合能。
在单散射理论架构中,χ(k)函数可表示为:
kχ(k) =(3)
该式为标准EXAFS公式。公式中,S02为振幅衰减因子,λ是光电子平均自由程,求和i为吸收原子周围不同配位壳层求和,Ai(k)是散射原子的振幅函数,φi是耦合吸附/散射原子的相函数,Ni是配位数,ri是吸收原子与散射原子之间的距离,σi是定量表示热电流壳层无序度的Debye-Waller系数。每个壳层,σi包括原子热运动(σi,T)的动态项和结构无序度(σi,D)的静态项,可表示为:
σi2 = σi,D2 + σi,T2 (4)
Sayers、Stern和Lytle首次提出EXAFS测得的Debye-Waller系数的双重内涵,并定量揭示了EXAFS中晶相和非晶相Ge的不同。公式4在催化研究中起着重要作用,因为无序度是材料中的重要参数。公式3在如下情况成立:(1)单散射路径在EXAFS信号中占主导;每个壳层键长的对分布函数可以用高斯函数e(-2k2σi2)表达。EXAFS标准公式(公式3)参数化了模拟吸收原子附近局域原子结构。EXAFS信号的震荡结构取决于吸收原子和散射原子之间的距离和光电子结合能Eb(公式2),它在sin(2kri)项中有所表达。EXAFS实验短程由指数项e(-2ri/λ)表示,它包括了核空穴的有限寿命和光电子有限平均自由程(λ)。干涉波强度通过背景散射振幅Ai(k)和配位数Ni取决于邻近原子的类型和数目。结构参数Ni,ri和σi2可通过最小二乘法获得。通过考察实验样品点kj,最小化实验和模型knχ(kj)函数的差异。最小化既可以在k空间完成,也可以通过Fourier变换函数R空间表达。对于每个配位壳层,配位数、原子间距和热力学系数都可以在EXAFS研究中获得。根据Nyquist原理,优化参数中的最大数目(nind)由k空间间隔(Δk)和R空间间隔(ΔR)定义,可以表示为:
nind = 2ΔkΔR/π(5)
图2.单重和多重原子散射示意图
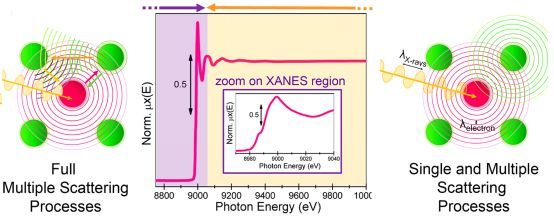
2.2 多重散射理论
标准EXAFS公式(公式3)仅考虑了单散射近似。它仅假定光电子的终态只受到近邻原子的单散射过程的影响。然而,如果光电子波在传播到吸收原子前被多个近邻原子多次散射,这样单散射近似将不能适用,而必须考虑多重散射的影响。尤其是对于多个个原子呈直线或近似直线排列的路径,其多重散射的贡献甚至会比高壳层单散射的贡献还大。Lee和Pendry首次对多重散射EXAFS作出理论论述,他们把多重散射的贡献按其有效路径长度进行分类。Rehr和Albers发展了基于格林函数传播子可分离表示的路径展开方法,克服了以往的多重散射理论计算方面的主要困难。
然而,尽管在多相催化中,单散射理论足够获得表面活性物种的结构和参数,然而在有些实例中,多重散射理论的贡献是很必要的,并应包括在EXAFS数据分析中。当讨论一个三体路径(一个吸附原子A和它的两个近邻原子B和C)时,除了经典的两体单散射贡献(A → B → A和A → C → A),也需要考虑到多重散射贡献(A→ B → C → A)。三体多重散射贡献正比于cos(θ),θ为A-B-C角。这表明原子共线排列(B-A-C或A-B-C)大大增强了多重散射贡献,称之为遮蔽效应。
多重散射理论的发展是现代XAFS理论成功的关键步骤。该理论可以同时处理EXAFS和XANES。关键问题是收敛性,即需要多少项,它们是什么。进一步研究表明低重或多重散射理论都不能令人满意。计算多重散射理论到更高阶主要有三种主要的计算困难:(1)需要高能的高角动量基底;(2)多重散射路径的指数性增加;(3)需要多重散射Debye-Waller系数。
3.EXAFS数据处理
基于单散射理论,处理EXAFS数据分析的代码有Michalowicz发展的EXAFS pour le Mac,Klementev发展的Viper,RSXAP,SEDEM,NPI和XAS。这些代码被广泛应用于χ(k)。过去几年,也有基于多重散射理论的GNXAS,EXCURVE和FEFF等。EXAFS标准的数据分析过程一般包括消除噪音、边前背景扣除、数据归一化、k空间转换、Fourier变换到R空间、反Fourier变换和曲线拟合七个步骤。
消除噪音。噪音是指由于单色器中不正常的反射信号或样品中的衍射信号。XAFS谱经常带有一些异常尖锐的假信号峰,这些尖峰甚至会比EXAFS信号的幅度强得多,会在Fourier变换谱上产生鬼峰。因此,需要在进一步数据处理前消除一些影响太大的假信号。消除方法是对假信号前后的数据做多项式拟合来内插假信号区域内的数据。
边前背景扣除。在分析EXAFS数据时,研究人员通常只关注吸收边后的数据,而对吸收边前的数据不甚关注,一般用Victoreen经验公式:
μν(E) = aE-3 + bE-4 (6)
或用修正后的Victoreen经验公式:
μν(E) = aE-3 + bE-4 + c(7)
得到边前吸收背景。拟合边前数据,再外推到边后的吸收背景。
数据归一化。这是指把不同样品测得的吸收系数统一到同一标准,来抵消因样品量差异所带来的影响,归一化公式为:
χ(E) = (8)
其中,Δμ(E0)为吸收边位置的“边阶”高度。边前和边后几百eV的数据用低次多项式分别进行拟合,外推到E0位置,两线在E0的差值是边阶Δμ(E0)。
k空间转换。E空间的EXAFS数据χ(E)通过:
k= [2m(E- E0)/E2]1/2 (9)
为了补偿信号在高k部分的衰减,χ(k)需要进行kn的加权。
Fourier变换到R空间。对经过kn加权的曲线作Fourier变换,把数据转化到R空间。通常EXAFS分析中设定Δk= 0.005 nm。从R空间谱中可得到近邻原子的径向分布以及各壳层近邻原子对XAFS数据的贡献。
反Fourier变换。将R空间中的配位壳层峰反变换回k空间,可以把指定壳层的振荡信号从原谱图中分离出来,提供拟合过程从中解出结构信息。
曲线拟合。将单一壳层的振荡信号分离出来后,可以用EXAFS基本公式(公式3)拟合出定量的结构参数。在拟合之前必须先获取振幅和相移函数,这些函数可以通过处理标准化合物的实测谱得到,或通过理论计算得到。
4.XANES数据处理
由于吸收边附近的光电子平均自由程远高于EXAFS区域发射出的光电子平均自由程,因此完整的多重散射理论是唯一可能分析XANES数据的方法。Frascati课题组首次发展出可应用的多重散射算法,CONTINUUM。其他算法还有FEFF-8、FEFF-9、FDMNES、Wien2k、PARATEC、BigDFT、SPRKKR、PY-LMTO-LSDA、XKDQ……
5.XAFS在多相催化中的应用
EXAFS对催化剂的研究起始于无负载析出相,这是因为它代表了活性负载相的模型材料。由于它们的非晶相性,EXAFS谱是判定吸收原子附近局域结构的唯一结构技术。图3显示了非负载相的EXAFS。Fourier第一壳层位于1.6 Å,代表其它壳层的更弱和更复杂位的峰位于更长的距离(两个峰位于2.7 Å和3.2Å)。Troitskii等基于Pd2+多核羟基络合物,提出拟合数据的结构模型。Pd2+的局域环境包括PdO4的平面协调方,PdO4单元有两个O1a或一个位于桥位的O1b连接,余下的Pd-O键连接了OH官能团。为了限制EXAFS拟合的自由参数,需要作出如下限制:三个Pd原子(Pd0,Pd1和Pd2)和六个氧原子(O1a,O1b,O1c及其等价物)被限制在同一平面上。因为没有显著的多重散射路径,包括Pd0和Pd1或Pd0和Pd2,几何结构近似不影响拟合结果。拟合数据的定量结果证明了Troitskii的结构模型,并且表示析出相的本质是[Pd(OH)2]n胶体,它具有高局域有序性和弱的长程有序。
图3.
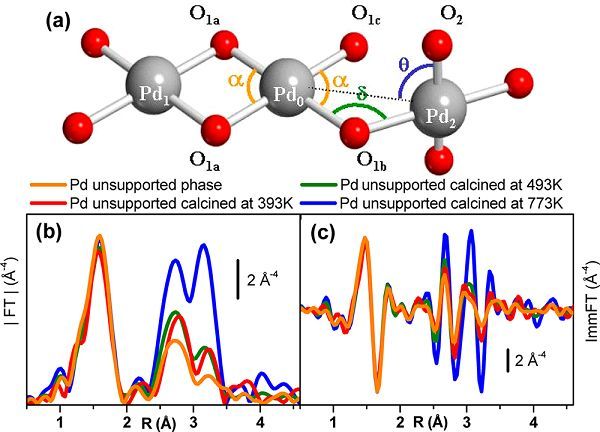
(a)Pd(OH)2理论模型;灰色为Pd原子,红色为O原子[Russ. Chem. Bull. 1995,44, 1822]。(b)R-空间Fourier变换模[Langmuir 2010, 26, 11204]。(c)R-空间Fourier变换虚数部分[Langmuir 2010, 26, 11204]。
XAFS是证明单原子催化剂最有利的证据,是球差电镜的完美补充。电镜只能给出局域信息,而XAFS给出统计平均概念。几乎是单原子催化剂必不可少的表征手段,见图4。
图4.
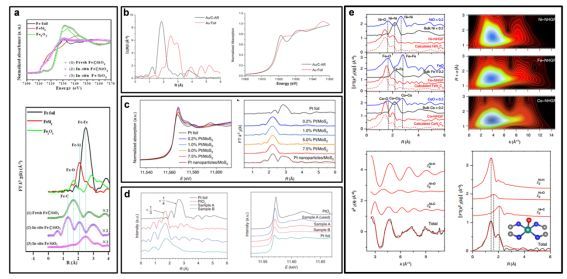
(a)0.5% Fe©SiO2 [Science, 2014, 344, 616-619]。(b)Au/C-AR[Science 2017, 355, 1399–1403]。(c)Pt/MoS2 [Nature Nanotechnol. 2018, 13, 411-417]。(d)Pt1/FeOx [Nature Chem. 2011, 3, 634-641]。(e)MN4C4(M=Co, Ni)[Nature Catal. 2018, 1, 63-72]。
6.EXAFS的优势及局限性
XAFS技术在多相催化中有重要应用,它具有如下优点:不依赖于晶体结构,可用于大量的非晶态材料的研究;不受其它元素的干扰,可对同一材料所含的不同的元素分别研究;;可测得配位原子的种类、配位数与吸收原子的间距等参数;;能够分析低浓度的样品,浓度可低到几个ppm。然而,XAFS也存在一定的局限性:由于结构和热无序的影响,难于得到5Å以上高配位壳层的信息;难以预测大颗粒金属原子团簇的形状。当原子周围相邻两壳层距离在0.5 Å或更小时,Fourier变换无法将其完全分开,采用多层拟合,其结果可靠性不一定使人满意此外,测得的结构参数是所有吸收原子的平均值,由于活性位的结构和形态多样化,无法完全鉴别催化反应活性中心。
其中,在XAFS技术中,研究人员普遍应用ΔXANES法于探测催化剂表面吸附物种的吸附位点。ΔXANES法是基于假设两个XANES谱中所有相同的贡献可以通过差分去掉,可表示为:
Δμ= μ(1) - μ(2) (10)
例如,对于负载型Pt基催化剂,吸附前后XANES谱μ分别为μ(1)和μ(2),其XANES谱差异可表示为Δμ,可以进一步表示为:
Δμ= μ(Ad/M) - μ(M) = Δμo + Δ(μo▪χM-M) + μoAd/M▪χM-Ad (11)
其中,μ(Ad/M)是吸附态金属催化剂的光谱,μ(M)是无吸附金属的光谱;Δμo等于由于吸附而改变的原子XAFS值;Δ(μo▪χM-M)是吸附物化学吸附引起的金属-金属散射的改变;μoAd/M▪χM-Ad是吸附物引起的散射。
科学指南针是互联网+科技服务平台,500多家检测机构,提供近5万种设备和服务项目,涵盖生物医药、智能硬件、化学化工等多个领域,由专业人员1对1跟踪服务,保证检测质量与效率。
免责声明:部分文章整合自网络,因内容庞杂无法联系到全部作者,如有侵权,请联系删除,我们会在第一时间予以答复,万分感谢。








