









 您已经拒绝加入团体
您已经拒绝加入团体

 2021-11-08
2021-11-08
 4285
4285
 0
0
【摘要】 通过光催化将水以2:1的化学计量比分解成氢气和氧气,太阳能驱动的制氢已经成为当前研究的焦点。

背景
通过光催化将水以2:1的化学计量比分解成氢气和氧气,太阳能驱动的制氢已经成为当前研究的焦点。为了在商业上与传统的化石制氢竞争,需要达到10%的能量转换效率,这就要求开发窄带隙光催化剂以高效收集太阳能。
为此,与氧化物光催化剂相比,金属硫化物尤其引人注目,这是由于硫的电负性较低,它们通常具有较窄的带隙。硫化物光催化剂总是发生由光生空穴引起的严重自光腐蚀,而不是通过水氧化生成O2。
迄今为止,使用硫化物基光催化剂进行化学计量水分解的成功率相当有限。如何实现有效的S2-的氧化−在硫化物中,光催化剂仍然是一项极具挑战性的任务。
研究思路与方案
Liu课题组报告了双缺陷硫化物光催化剂上以化学计量H2和O2的生成,该催化剂通过二维(2D)ZnIn2S4单层与Ag之间的简单水性阳离子交换反应获得。
如图1所示,阳离子交换过程产生了两种缺陷,即Ag掺杂剂和纳米孔。这种独特的结构提供了加速的表面反应动力学,这源于Ag掺杂促进析氧反应(OER),纳米孔处悬挂的S原子对析氢反应(HER)的改善协同作用。
因此,这种窄带隙硫化物光催化剂在没有任何助催化剂和异质结辅助的情况下表现出良好的活性和稳定性。
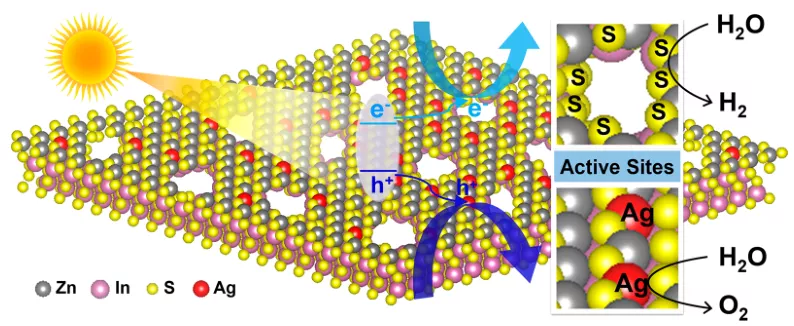
图1复合材料制备示意图。
结果与讨论
结构表征:
从图2a可以看出,水热制备的ZnIn2S4具有超薄结构。所得Ag-ZnIn2S4整体上继承了原始ZnIn2S4的形态,同时呈现出大量纳米孔(尺寸约为几纳米),均匀分布在整个薄片上(图2c)。
图2d显示这些纳米孔穿透单层,产生大量新暴露的活性边缘。图2e测得的面间距离和相应的二面角表示六方晶系的ZnIn2S4平面的[001]方向。图2f中的放大图像直接揭示了部分Zn原子被Ag原子取代。

图2(a)原始ZnIn2S4和(b)Ag-ZnIn2S4的TEM图像,(c)HAADF-STEM图和(d-f)Ag-ZnIn2S4的像差校正HAADF-STEM图,图f是与e中红线框包围的区域相对应的放大图像。
催化性能测试:
如图3a,Ag掺杂剂的最佳浓度为0.61 mol%时,Ag-ZnIn2S4的析氢速率达到7.3 mmol g-1 h-1,比原始ZnIn2S4(1.3 mmol g-1 h-1)。
此外,在56h的累积可见光照射下,Ag-ZnIn2S4的析氢活性在7个连续循环中非常稳定(图3a)。
可见光驱动的OER进一步在含有NaIO3作为电子牺牲试剂的水溶液中进行(图3b)。
相反,在相同的反应条件下,未掺杂ZnIn2S4光催化剂上未检测到O2析出。
值得注意的是,如图3c所示,在可见光照射下(λ>420 nm),在Ag-ZnIn2S4上稳定地产生H2和O2,其对应的速率可达56.6和29.1 umol g-1 h-1,使H2/O2比非常接近全水分解的预期2:1化学计量比。
此外,如图3d所示,Ag-ZnIn2S4在405、420和450 nm处的表观量子效率(AQE)别为0.70%,0.57%,0.20%。
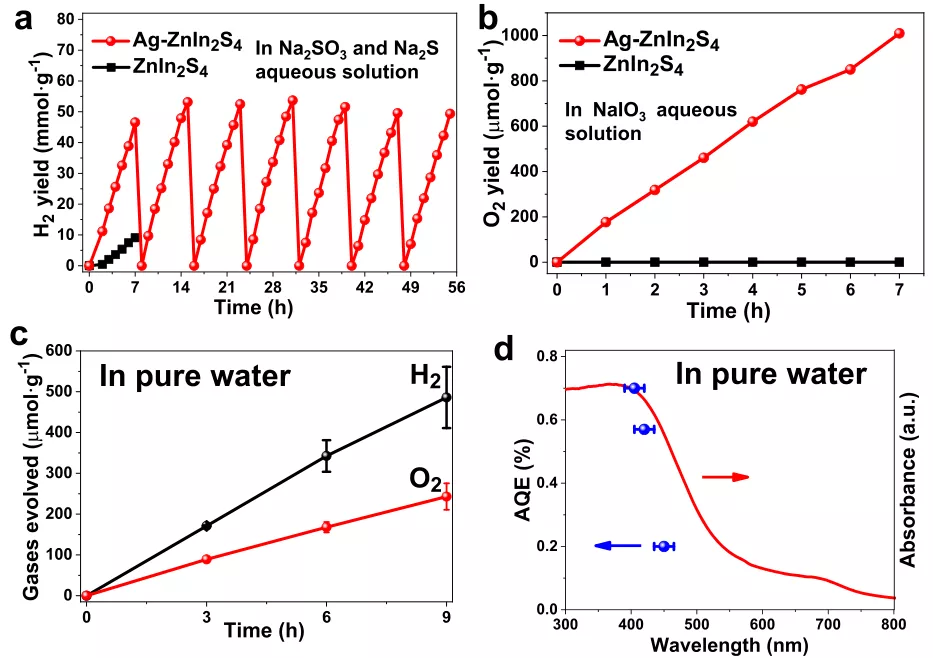
图3在可见光照明下,在牺牲试剂(HER为0.25 M Na2SO3和0.35 M Na2S水溶液,OER为20 mM NaIO3水溶液)存在下,ZnIn2S4和Ag-ZnIn2S4(a)H2半反应和(b)O2半反应的光催化性能比较(300 W氙灯,λ>420 nm),(c)在可见光照明下,蒸馏水中Ag-ZnIn2S4上全水分解的时间过程(300 W氙灯,λ>420 nm),(d)Ag-ZnIn2S4在光催化整体水分解中AQE的波长依赖性。
从图可以看出,作者通过Ag掺杂的制备方法,制备了Cu掺杂的ZnIn2S4,发现并不具有产生氧气的能力,如图4a所示。价带边缘电荷密度的空间分布表明,Ag掺杂剂和相邻S原子周围的电荷密度显著增加(图4b,e)。
在这种情况下,电子很容易被光激发到导带,并导致Ag附近的电子耗尽(图4c,d)。Ag掺杂剂更有利于H2O分子吸附,在Ag-ZnIn2S4上的能量为(-2.07 eV),而原始ZnIn2S4和Cu-ZnIn2S4的吸附能为-0.54(在Zn原子上)和-0.32 eV(Cu原子上)(图4f)。
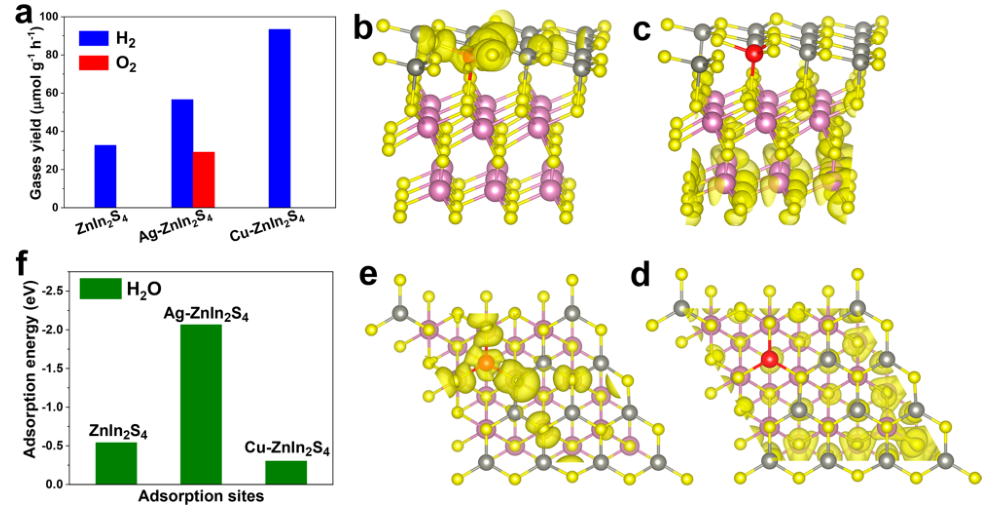
图4(a)比较ZnIn2S4、Ag-ZnIn2S4和Cu-ZnIn2S4在可见光照明(300 W氙灯,λ>420 nm)下光催化水分解反应中的H2和O2析出活性,(b,c,e和d)Ag-ZnIn2S4的(b和e)价带和(c和d)导带边缘附近的部分电荷密度分布,(b和c)和(d和e)分别为[001]方向的侧视图和俯视图。红色、灰色、紫色和黄色的球分别代表Ag、Zn、In和S原子(f)计算的H2O分子在ZnIn2S4、Ag-ZnIn2S4和Cu-ZnIn2S4上的吸附能比较。
本文所有内容文字、图片和音视频资料,版权均属科学指南针网站所有,任何媒体、网站或个人未经本网协议授权不得以链接、转贴、截图等任何方式转载。








