









 您已经拒绝加入团体
您已经拒绝加入团体

 2024-06-20
2024-06-20
 6769
6769
 0
0
【摘要】 6月16日,由科学指南针主办的第二届多尺度材料计算模拟国际研讨会在广州圆满落幕。

中国化学会第34届学术年会
多尺度材料计算模拟论坛
6月16日,由科学指南针主办的第二届多尺度材料计算模拟国际研讨会在广州圆满落幕。来自国内外200多位专家学者共聚一堂,从计算方法的突破和创新、计算预测与实验合成的有效结合、机器学习对计算的影响等多个维度,深入探讨了计算化学在不同材料领域应用的最新研究成果。带来了一场精彩纷呈的学术盛宴。下面,小编将采撷与会专家学者的精彩观点,以飨读者。
大会报告观点集锦

报告题目:精确高效的催化反应体系多尺度模拟
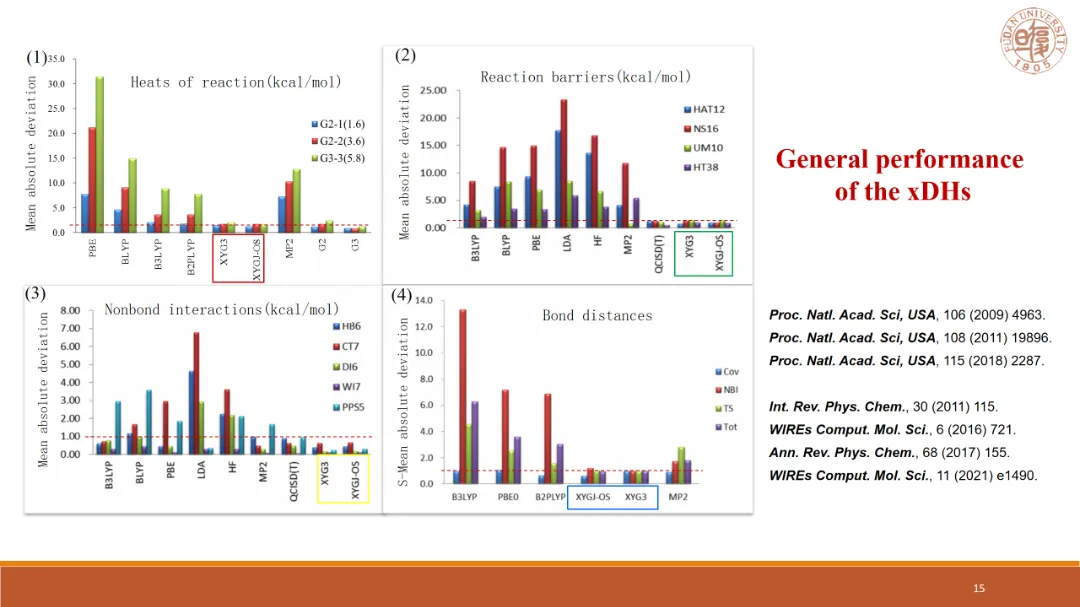
基于多尺度理论建模来探究界面纳米现象和纳米结构是一种良策,目前分子水平上主要有簇、嵌入式簇和周期性三种广泛使用的模型。每种模型都有其优势和局限性,因此最佳选择应基于待研究的催化剂的结构性质和电子性质来决定。这样以来,获取更为精准的相关性质就尤为重要。基于此,该报告从扩展的ONIOM(XO)模型入手,结合与XYG3型双杂化泛函结合,对不同理论水平上处理的模型,包括使用周期性边界条件来研究固体表面进行了研究。并在反应热、过渡态、各种相互作用以及几何结构的描述等方向有很大的进步,可以有效控制误差,为具有不同成键性质的催化剂提供准确而高效的模拟。

报告题目:二维材料计算设计的若干成功实例
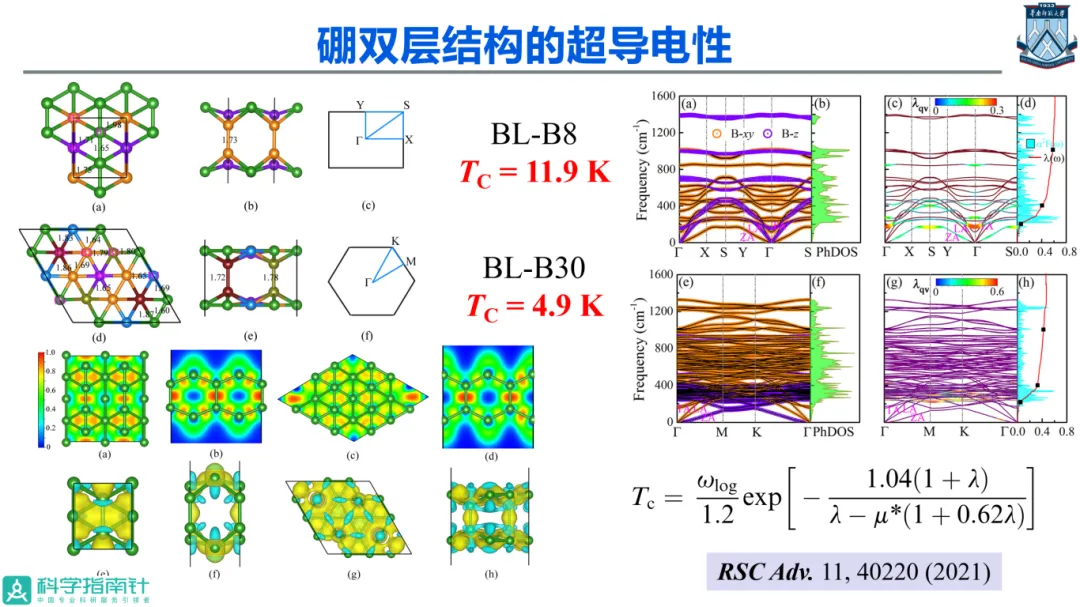
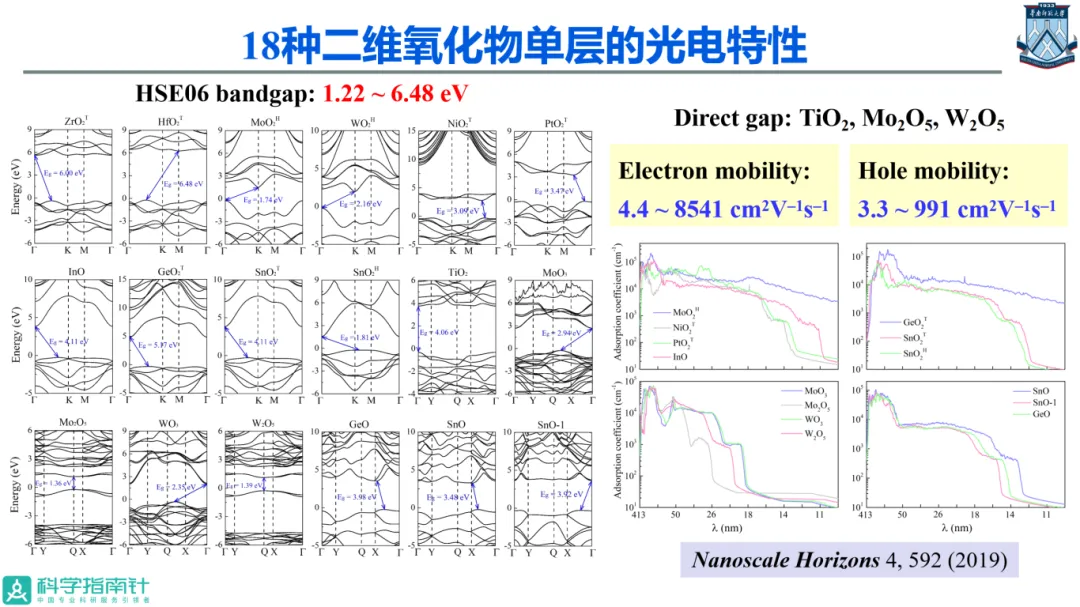
在二维材料研究领域,通过第一性原理模拟软件和高性能计算机进行材料设计与性能预测,预测新的现象和效应,提供原子级的物理图像,对于推动该方向材料研究发挥着重要作用。
该报告通过计算设计具有高环境稳定性的二维氧化物和二维磁体,如CrSBr磁体、MBene、硼烯双层、InO单层、AgCr2S4和AgCr2Se4二维多铁体系等,预测了其电子能带结构和物理性质,同时得到了实验的若干成功验证,有助于拓展二维材料在信息、能源等领域的应用。基于此,可以期待计算材料学在未来的低维材料研究中发挥更积极的做,其关键在于从实验和器件应用中寻找出有意义的科学问题,充分发挥计算模拟的优势。

报告题目:氢能催化材料的理论与实验研究
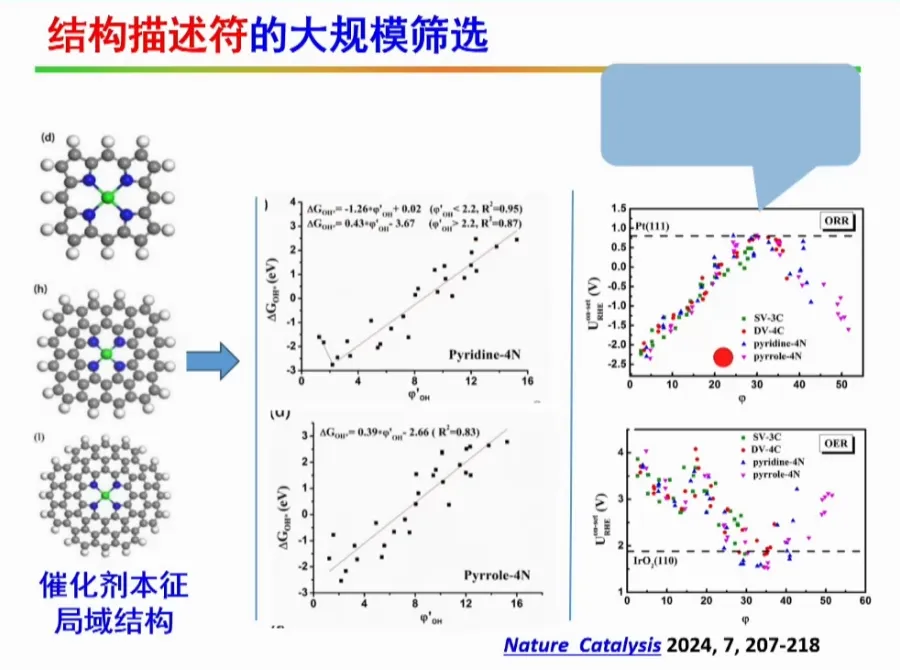
针对氢能的制造和应用,发展高效的催化剂成为了关键。活性位点原子级分散的催化剂由于其原子利用率可达100%,近年来已受到了广泛关注。
本报告提出制备原子级分散催化剂的通用制备方法,成功制备出系列催化剂,并首次揭示了单原子锌/铁催化剂局域结构配位是其决定其催化活性的关键,设计制备了轴线氧掺杂的铁基催化剂。同时大规模筛选了双原子催化剂,针对特定的双原子催化剂(如Fe-Cu,Fe-Mn双原子催化剂),实现了其靶向合成及其在氢燃料电池的应用,并提出了双位点共吸附拟制双氧水产生的催化设计新方法,为设计高性能燃料电池催化剂提供了新思路。
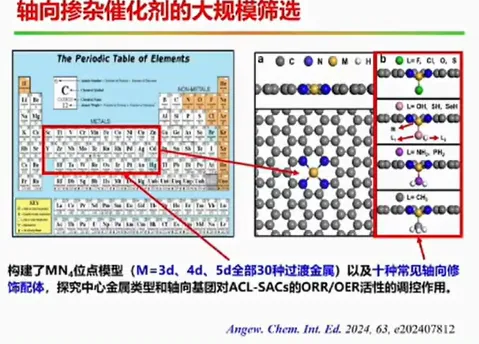
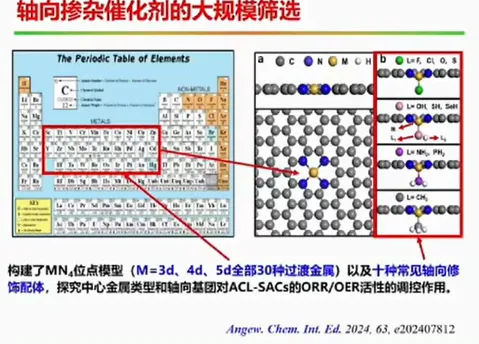
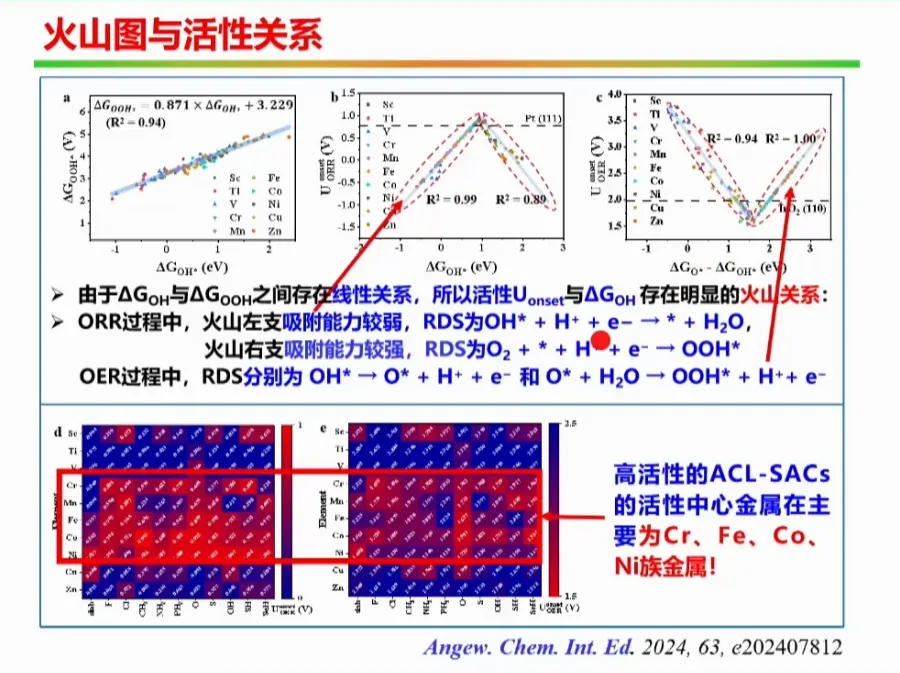

报告题目:电势影响下的单原子催化剂电子结构研究
经典的计算氢电极方法广泛应用于传统电催化领域。该方法的核心内涵在于保持体系电子数恒定(恒电荷方法)。实际的电极材料会因为外加电极电势带有净电荷,影响电极材料的表面物理化学性质,因此应当通过适当方式保持体系电势而非电荷的恒定。
动态调整体系电荷量是实现恒电势计算的一种重要方法。对于典型的石墨烯基碳氮载体负载单原子催化剂而言,恒电势下催化剂材料的过渡金属单原子d轨道的电子占据数处于动态变化中,继而显著影响电催化反应中间体的吸附能,调控电催化反应活性。恒电势方法基于对催化剂微观的电子结构的动态调控显著改进了反应能的计算精度,引领了电催化理论计算的范式革命。
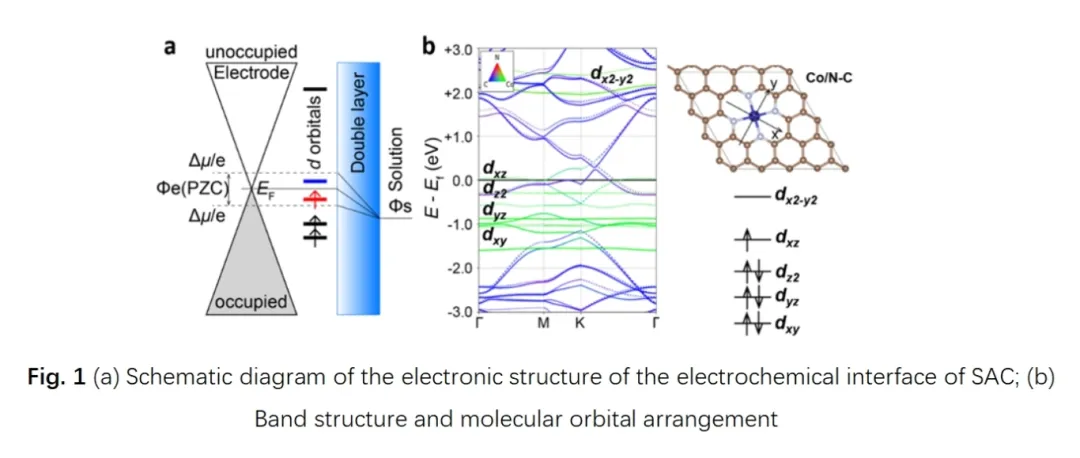

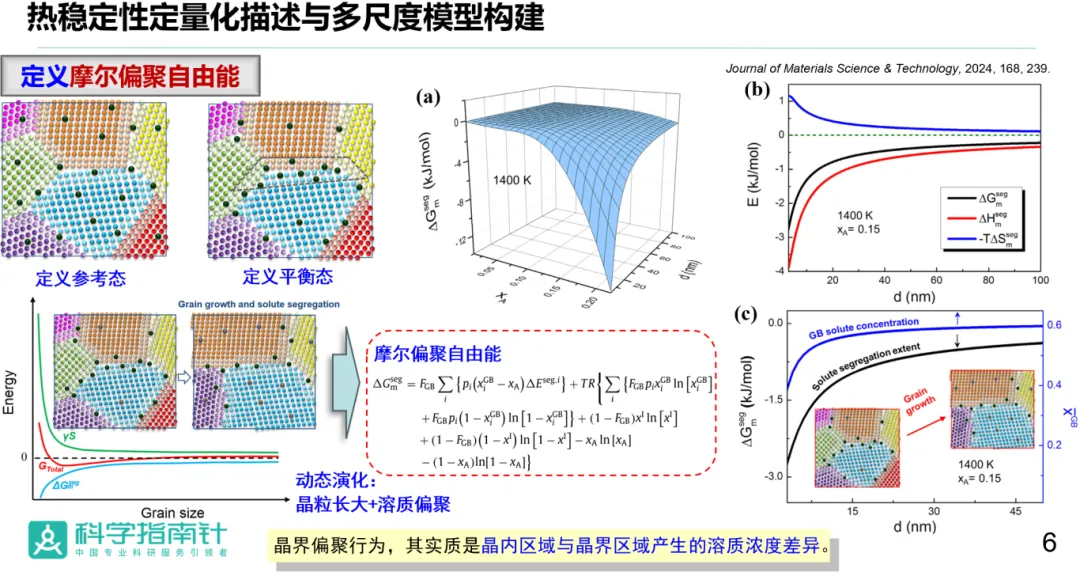
报告题目:纳米合金中溶质偏聚与热稳定性的多尺度模型研究
纳米结构合金材料的热稳定性问题是阻碍该类材料广泛应用的瓶颈之一。以往研究表明,添加合适的溶质元素,通过晶界偏聚效应,可使界面能量有效降低,从而增强纳米合金体系的晶粒组织稳定性。但是,长期以来对于溶质元素与基体的微观相互作用并未明确,关于掺杂元素的筛选及其对纳米晶合金界面的调控机制缺乏系统的理论研究。
基于以上原因,唐老师围绕纳米晶固溶体材料的晶粒组织稳定性构建了多尺度模型,引入摩尔偏聚自由能概念来量化封闭体系中溶质元素的实际偏聚能力,描述了界面结构、晶粒尺寸、材料成分和外界温度等关键参量对偏聚行为的影响。基此阐明了溶质偏聚与晶粒生长的交互作用,进而探讨了溶质偏聚对纳米晶结构热稳定的影响机制。本工作提出了纳米结构失稳的新热力学判据,认为摩尔偏聚自由能和晶界总能随晶粒尺寸的变化速率的竞争关系是体系能否稳定的关键。该研究为实现固溶体合金中纳米结构的高温稳定化提供了理论指导。

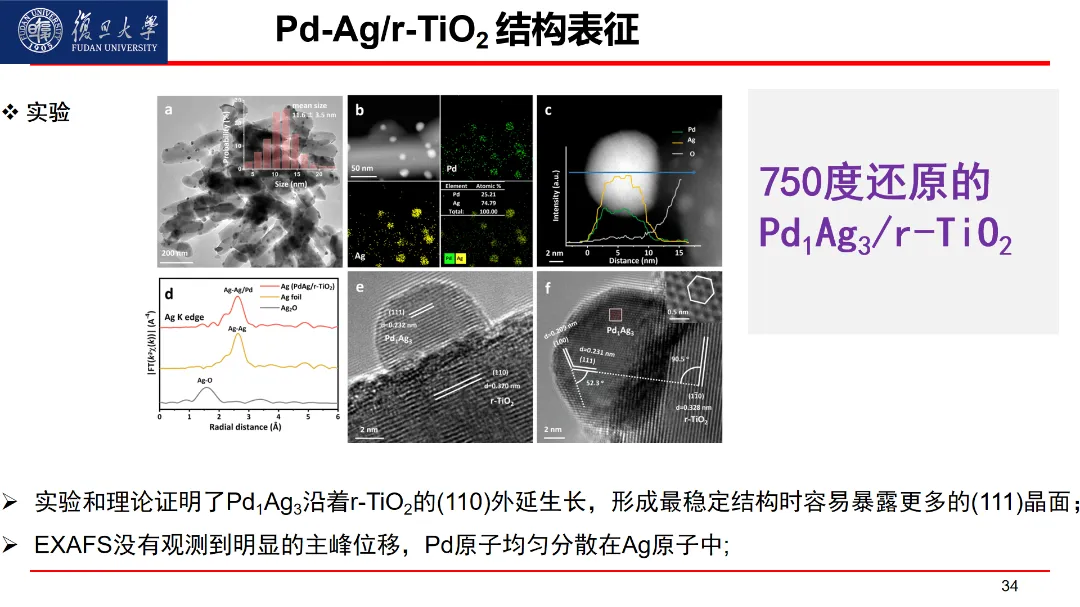
报告题目:PdAg催化剂乙炔选择性加氢理论和实验研究
乙炔半氢化反应是制备高纯度聚合级乙烯的关键步骤,在石油化工领域中具有显著的经济意义。尽管工业上广泛采用金属钯(Pd)作为活性成分,但纯 Pd 容易引起过度氢化和积碳,这会降低对乙烯的选择性和催化剂的稳定性。因此,探究Pd 在乙炔半氢化反应中的催化本质,揭示银(Ag)在工业常用的 PdAg 体系中催化剂的结构演变及其对催化性能的作用,以及开发可替代工业应用的新型催化剂,是科研界与工业界共同关注的焦点。该报告详细介绍了通过将LASP人工智能原子模拟和实验相互指引结合的方法,对Pd和PdAg体系乙炔半氢化反应选择性调控研究的结果。实验发现Pd1Ag3/HT 催化剂在60 ℃时,乙烯的选择性在乙炔转化率大于96 %时能长时间保持在80 %以上;通过高通量实验数据的随机森林分析与预测,结合机器学习原子模拟方法,开发了一种具备卓越选择性和稳定性新型催化剂:负载在金红石二氧化钛 (r-TiO2)上的Pd1Ag3合金催化剂。

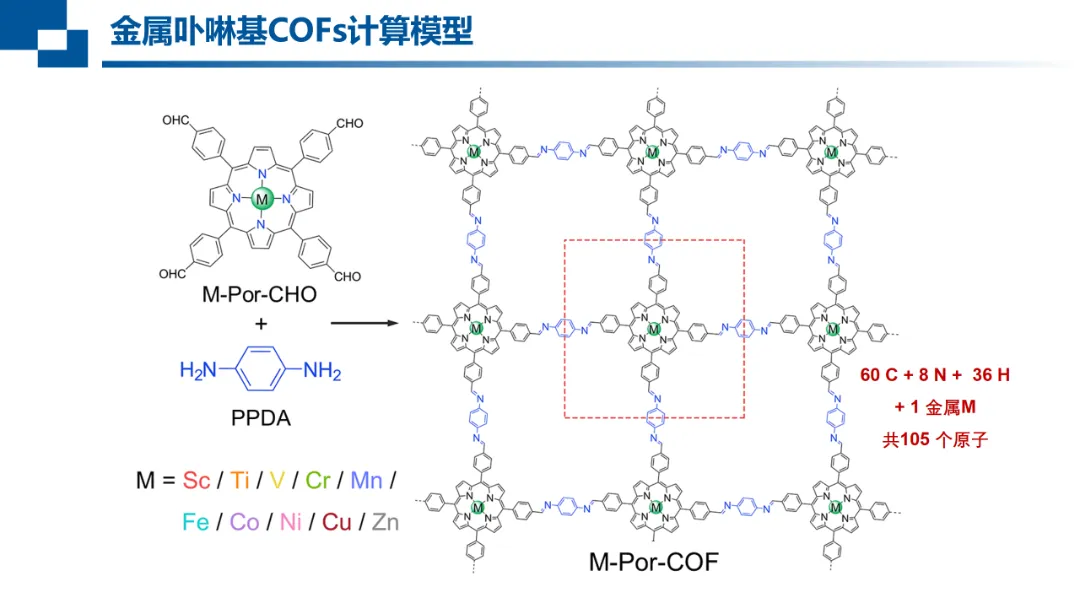
报告题目:共价有机框架材料催化CO2电还原的理论研究
金属卟啉基共价有机框架材料作为新兴的重要单原子催化剂,因其具有结构可调,易功能化和高比表面积等特点,有潜力成为二氧化碳还原反应的高效催化剂。通过对过渡金属配位环境的调控能够极大丰富催化剂结构类型,有望从中筛选有前景的新型催化材料。
通过对框架载体的设计(C、N混杂和O、S杂原子取代)和3d过渡金属的筛选,预测了一批具有潜力的催化剂。提出以上配位调控策略通过促进催化剂向被吸附物的电子转移,稳定与电位决速步相关的中间体,从而提高电催化还原活性。进一步针对杂原子取代卟啉的CO2还原为CO反应网络的探索,发现该过程遵循电子转移与质子转移交替进行的分步机理,增进了对反应完整机理的认识。

报告题目:多孔光催化材料理论设计
相比传统的光催化材料,多孔金属有机框架(MOF)和共价有机框架(COF)材料具有一些独特的特点和优势,如结构丰富、可后修饰、孔径可调等。随着近年来对MOF和COF材料研究的不断深入,这两类材料已然成为两类重要的光催化材料。光催化材料通过吸收光子产生光生电子和空穴,产生的载流子能够驱动氧化还原反应,进而将光能转化为化学能。将光生电子和空穴进行有效分离能够显著增加载流子寿命,从而提升材料光催化性能。在本次报告中,分别介绍了MOF和COF光催化材料电荷分离性质的调控策略。对于MOF,可以通过调控金属(Node)或配体(Linker)的结构增强配体-金属电荷转移(Ligand-to-Metal Charge Transfer),进而提升电子空穴对的分离效率。对于COF,可以利用N原子或者NH2等基团修饰来破坏结构的对称性,从而达到提升电荷分离效率。最后,针对多孔材料,简要介绍了QM/MM多尺度模拟方法和程序。该方法计算效率高,并在能量、结构、电子性质等方面的计算中均表现优异。

报告题目:锂离子电池材料的理论设计与优化
锂离子电池层状正极材料具有重要的应用价值。在高脱锂状态下,层状结构演化剧烈,结构稳定性以及离子扩散过程错综复杂,涉及到多种缺陷的产生和副反应的发生,例如阳离子混排,氧空位形成,气体析出,过渡金属溶解,以及电解液分解等。卢老师针对层状材料的基本问题进行了归纳总结,紧密围绕晶格氧展开了系列研究工作,尤其在晶格氧活性的触发机制、利用晶格氧活性的措施等方面取得了重要进展。卢老师从配位环境和分子轨道的角度,确定了阳离子混排诱导的电子结构重组的基本形式,以及产生这种现象的本质原因。在高脱锂态下,这两种电子状态之间可以发生简并和耦合,导致额外晶格氧活性的激发,以及过渡金属氧化还原的抑制,属于一种能量激发的状态。对于结构可逆性,这种电子重组增加了体系中产生氧二聚体或者氧气的可能性,并且建立了阳离子混排和氧流失之间互相促进的热力学关系。研究结果表明,电子重组是传统层状材料发生严重退化的一个关键的步骤和节点。通过调控能带结构,电子结构的重组过程可以得到调节和规范,从而有利于提升材料的电化学稳定性。卢老师的研究工作为锂离子电池层状正极材料的设计研发提供了重要的理论依据。
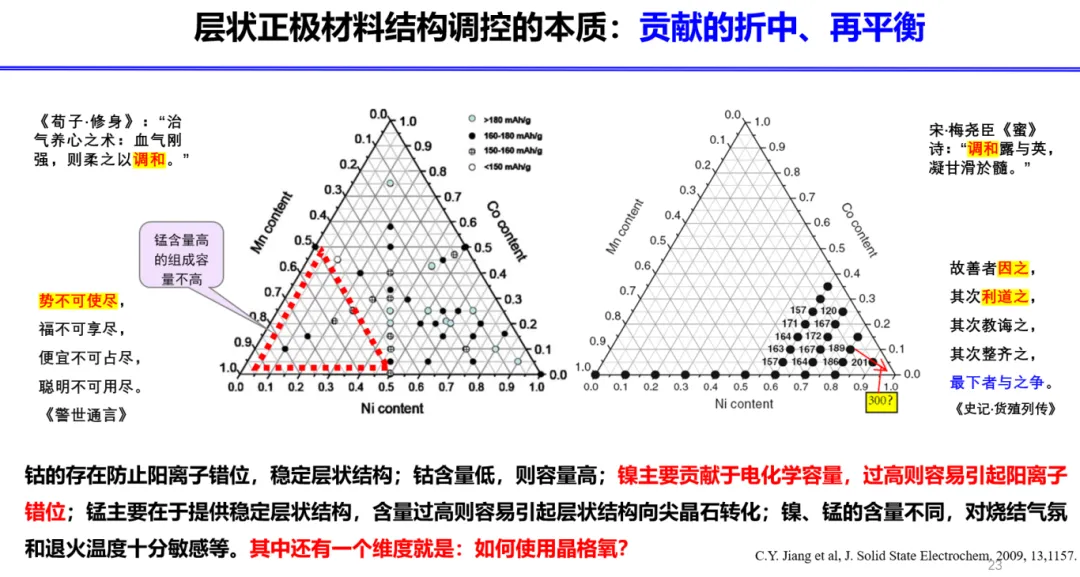

报告题目:材料表界面智能理性计算
在研究材料的过程中,会因为多尺度,多时间让材料的研究受到了一定程度的限制。针对这些问题,展开了一系列理论框架的研究。
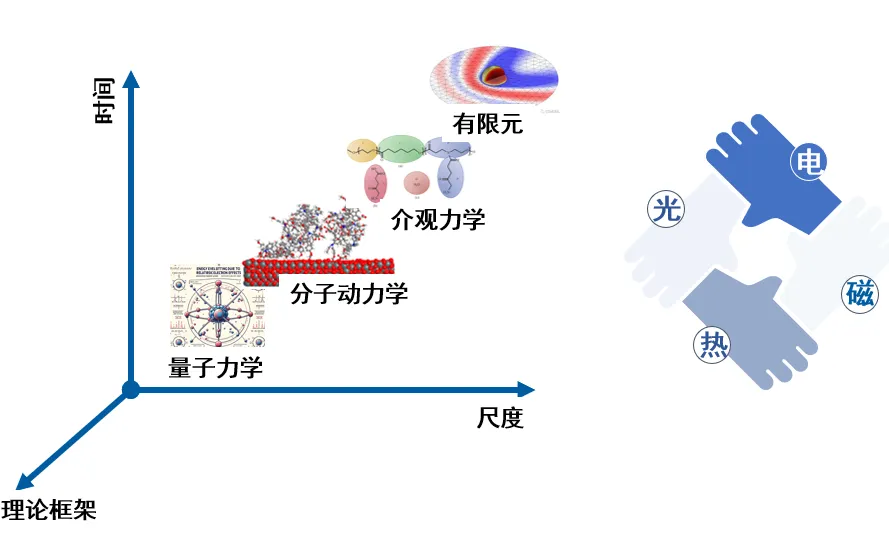
本次报告,详细介绍了报告人在材料表界面与人工智能领域的研究成果,研究涵盖了材料化学理性计算与仿生智能预测。首先,在理性计算方面,研究者重点研究了表界面光、电、热过程的协同模拟、稀土功能配合物的全电子相对论模拟及分子电场和晶格电场的理论计算。通过量子力学、分子动力学、介观力学及有限元等方法,成功提升了紫外光稳定性和界面稳定性,并揭示了Pb簇表界面的金属传输性和金属卟啉电荷转移性质。
在智能预测方面,开发了聚集态结构智能仿生搜索、晶体结构智能精确预测及基于图神经网络的高效预测方法,显著提高了分子和晶体结构的预测精度。通过遗传算法和卵型势驱动高效聚集态搜索实现从原子球密堆积到分子表面拓扑的转变,成功预测酞菁晶体结构。

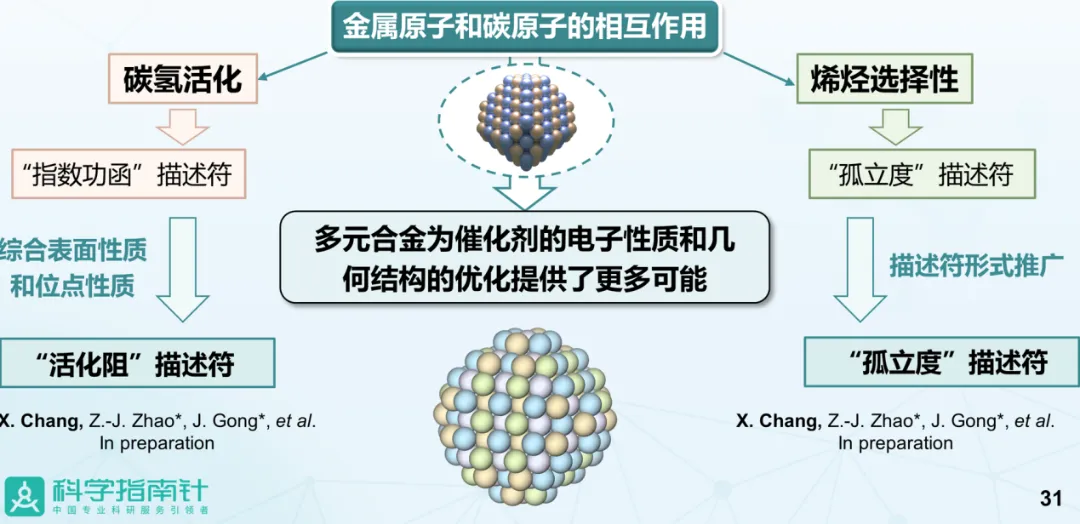
报告题目:烷烃脱氢合金催化剂的理性设计
丙烯是一种重要的化工原料。高效催化剂的制备是丙烷脱氢技术的关键,理论计算相比实验试错,可以起到加速催化剂设计的作用。但是,合金中催化位点的微观环境描述,计算描述符与催化性能的关联度等方面,依然充满挑战。此外,丙烷脱氢反应中目标产物丙烯的选择性由丙烯脱附和丙烯进一步脱氢这两个变量共同决定,难以直接预测。基于上述原因,常老师针对单位点合金,提出“催化微环境预测催化性能”的研究策略,发展了基于位点微环境直接预测催化性能的“孤立度”描述符。该研究具体采用了“将这二者解耦,分别定量化描述”的思路,以“吸附质和活性位的排斥作用”为纽带,利用简单的数学形式将这二者的定量描述耦合起来,从而凝练出微环境描述符“孤立度”。进一步的,常老师引入指数功函数描述符用以评估合金碳氢键活化,该描述相比d带中心,可统一描述M-C排斥和碳氢键活化差异,结合实验验证,发现其对碳氢键活化能力具有良好的描述效果。常老师的本项研究工作对推动催化剂从实验试错向理性设计转变提供了重要的理论支撑。
此次论坛内容精彩纷呈,汇聚了众多专家智库资源,获得了参会学者的高度认可。未来,科学指南针将在科研领域持续深耕,朝着“世界级科研服务机构”的目标努力,为科研工作者带来更专业、更高质量的学术交流活动,为科学发展和技术创新做出更大贡献。








