









 您已经拒绝加入团体
您已经拒绝加入团体

 2026-05-09
2026-05-09
 516
516
 0
0
【摘要】 科学指南针提供 Li‑Si‑C 三元体系 NEP 势函数定制服务,融合 DFT 与机器学习方法,适配硅碳负极大规模 MD 模拟、体积变化分析与微观机理解析。

硅碳负极凭借较高理论容量,成为锂电池下一代关键候选材料,体积膨胀现象一直是行业研发关注的重点。传统 DFT 计算具备良好精度,却难以支撑大规模原子模拟;通用机器学习势函数在复杂体系中容易出现精度偏差。依托顶刊相关研究成果,科学指南针推出 Li‑Si‑C 三元体系神经演化势函数(NEP) 定制服务,在 Li‑Si‑C 体系模拟场景中体现出较高精度与计算效率,为硅碳负极理性设计提供微观模拟依据。
一、硅碳负极模拟的核心技术痛点
DFT 计算可输出可靠的能量与力场数据,但面对数十万原子的 Si@C 核壳界面体系,计算效率偏低,难以开展长时程 AIMD 分子动力学模拟。常规机器学习势函数泛化能力有限,在复杂界面、结构缺陷、高温工况下容易出现精度波动,较难匹配科研与工业研发的模拟需求。
二、Li‑Si‑C NEP 模型核心技术特点
结合现有研究框架构建的 神经演化势函数(NEP) 模型,在模拟精度与计算效率层面具备明显优势,核心特点如下:
高效直接采样策略
仅选取原始数据池中 4.3% 的结构样本,共计 3114 个结构,便可覆盖 90% 以上 的结构特征空间,有效降低数据标注与模型训练成本。
主动学习迭代训练
模型可自主识别高不确定性构型并开展定向优化训练,能量预测误差可达 21.2 meV/atom,能够较好复现 DFT 计算得到的径向分布函数与锂离子扩散系数。
超高速模拟能力
整体计算速度相对传统 AIMD 提升约 7 万倍,可适配大规模原子级模拟场景,有效缩减模拟运算周期。
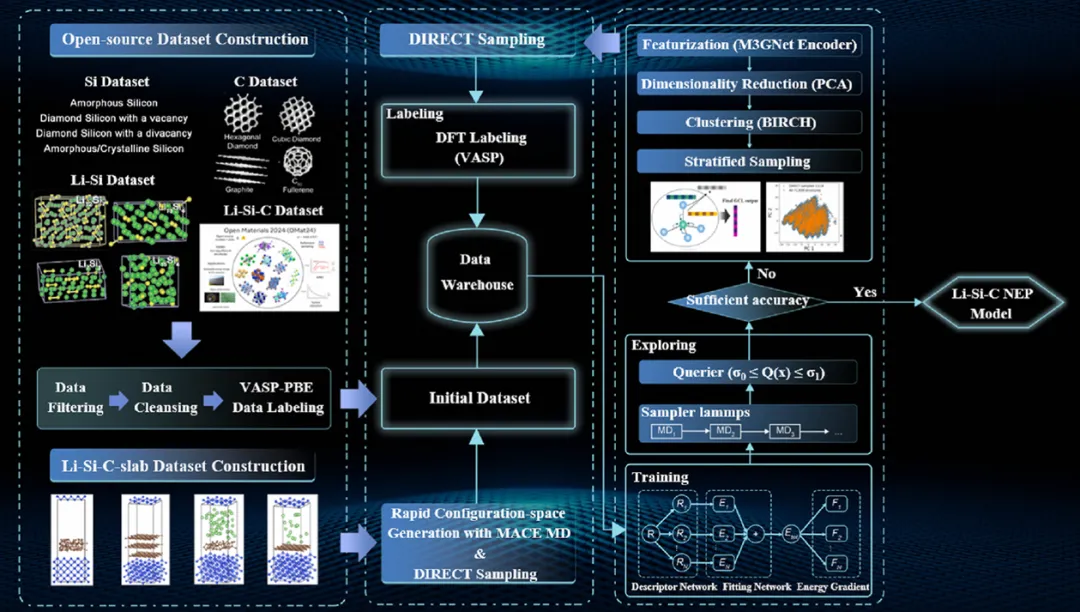
图1:用于设计 Li−Si−C NEP 力场模型的示意工作流程
三、Li‑Si‑C NEP 模型标准构建流程
科学指南针可围绕具体材料体系开展 DFT 标注、主动学习训练和模型验证,采用标准化流程搭建 Li‑Si‑C NEP 模型,保障模型可靠性与场景适配性:
1.数据集构建:完成 Si、C、Li‑Si、Li‑Si‑C 多类型结构数据采集整理;
2.特征化与降维:采用 M3GNet 编码器进行结构特征化处理,搭配 PCA 算法完成维度缩减;
3.聚类与标注:通过 BIRCH 聚类划分结构类型,依托 VASP/DFT 完成高精度能量与力场标注;
4.分层采样与训练:采用 DIRECT 分层采样方式,结合主动学习策略完成 NEP 模型训练与多维度验证。
四、机器学习势函数相关服务内容
全流程势函数定制开发
涵盖结构采样、DFT 数据标注、主动学习训练到多维度精度验证全链条,提供完整的机器学习势函数开发服务。
势函数精度提升与盲区修补
针对通用势函数在复杂界面、高应变、高温场景下的精度漂移问题,通过定向强化训练完成性能优化,将误差控制在 meV 级别区间。
高通量模拟与微观机理解析
支持万级至十万级原子体系 大规模 MD 模拟,可计算扩散能垒、体积变化、应力分布等关键参数,从原子层面辅助解析材料性能演变机制。
五、技术优势与适用对象
核心技术优势
-
数据成本可控:模型训练集规模可缩减 95% 以上;
-
误差表现稳定:能量误差可控制在 < 25 meV/atom,力误差 < 0.15 eV/Å,达到原文报道的验证水平;
-
工况适配范围广:可适配 5–20% 拉伸应变、500–2000 K 温度区间模拟;
-
大体系运算支持:适配万级至十万级原子体系并行计算;
-
体系可迁移性强:可延伸应用于金属、氧化物、电极界面、电解液等多种材料体系。
适用服务对象
高校科研课题组、电池材料企业研发部门、具备内部模拟计算需求的科研团队。
六、总结
依托机器学习势函数与 DFT 融合技术思路,Li‑Si‑C 三元体系 NEP 模型可为硅碳负极体积膨胀机制解析提供高效模拟支撑。结合直接采样与主动学习技术,能够在控制数据成本的同时,兼顾模拟精度与运算效率。科学指南针可提供专业的机器学习势函数开发服务,为硅碳负极材料机理研究与结构设计提供可靠的计算模拟支撑。








