









 您已经拒绝加入团体
您已经拒绝加入团体

 2024-09-12
2024-09-12
 926
926
 0
0
【摘要】 电化学技术在先进材料研究中的应用:原位FTIR、拉曼、XRD与DEMS分析
1.原位电化学红外

文章名称:Unveiling synergy of strain and ligand effects in metallic aerogel for electrocatalytic polyethylene terephthalate upcycling
文章链接:https://doi.org/10.1073/pnas.2318853121
期刊信息:1区;IF: 12.779;Proceedings of the National Academy of Sciences;
科学指南针测试服务:
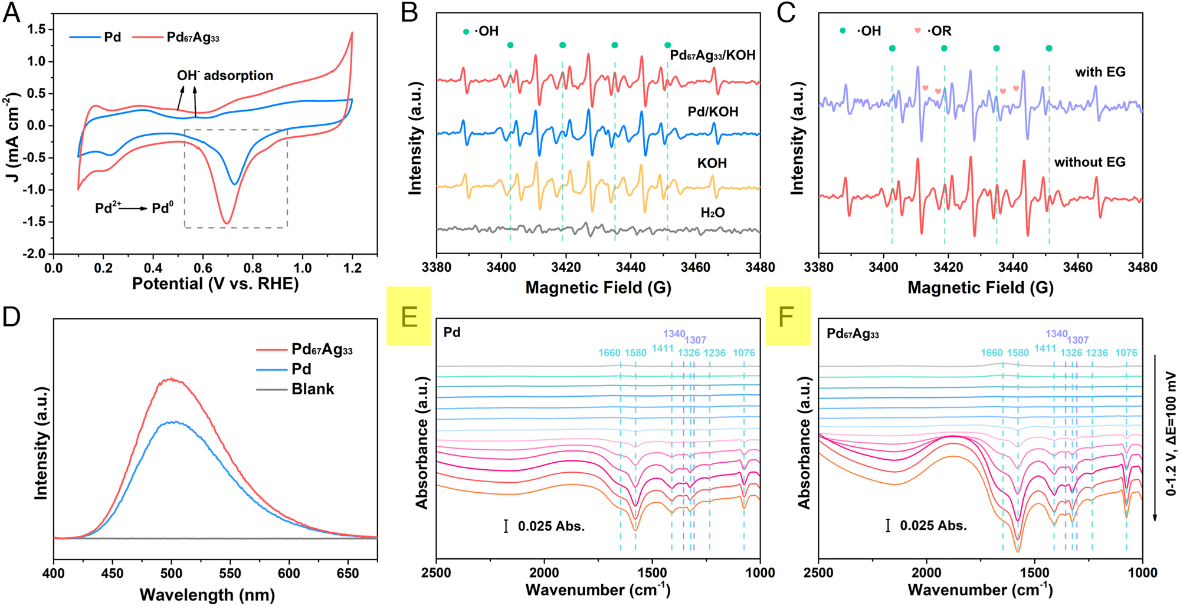
图4: 催化剂上中间体的选择性吸附和转化。(A) Pd和Pd67Ag33气凝胶在1 M KOH下的CV曲线;(B)和(C)使用DMPO作为捕集剂的电解质的EPR光谱;(B) OER和(C) EGOR;(D)使用香豆素(0.2 mM)指示剂检测电解质中·OH自由基的荧光光谱;(E和F) Pd和Pd67Ag33在EGOR操作下的原位电化学FTIR光谱
为了在分子水平上深入了解EG - GA转化过程中的反应机理,文中通过在科学指南针测试机构进行了电化学原位FTIR反射光谱检测,利用配备液氮冷却型碲化汞镉探测器(MCT-A)的Thermo 8700光谱仪收集了原位FTIR光谱。如图4 E和F所示, Pd67Ag33上的1,076和1,580 cm−1波段出现在0.5 V vs. RHE下,低于Pd (0.6 V vs. RHE),表明Pd67Ag33在EG - GA转换中表现出减弱的能量势垒。同时,Pd和Pd67Ag33的原位FTIR光谱比较发现,GA(1,076、1,236、1,326、1,411和1,580 cm−1)在Pd67Ag33上的信号显著增强,而不良产物相关波段(1,307和1,340 cm−1)的强度没有明显变化。这一结果进一步阐明了Pd67Ag33上GA选择性的提高,与NMR结果一致。
文章摘要:
在Junliang Chen等人的文章中报告了一种可持续的电催化方法,允许在环境条件下使用Pd67Ag33合金催化剂从废聚对苯二甲酸乙二醇酯(PET)选择性合成乙醇酸(GA)。
研究揭示了合金催化剂中应变和配体效应的协同作用,为未来PET升级回收催化剂的设计提供了指导。文章中进一步研究了Pd67Ag33催化剂在CO2还原反应(CO2RR)中的多功能性,并组装了EGOR//CO2RR集成电解槽,为将废碳资源(即PET和CO2)转化为高价值化学品提供了开创性的示范。
2 原位电化学拉曼

文章名称:Improving the Oxygen Evolution Activity of Layered Double-Hydroxide via Erbium-Induced Electronic Engineering
文章链接:https://doi.org/10.1002/smll.202206531
期刊信息:1区;IF: 15.153;Small, 2023, 19(5): 2206531.
科学指南针测试服务:

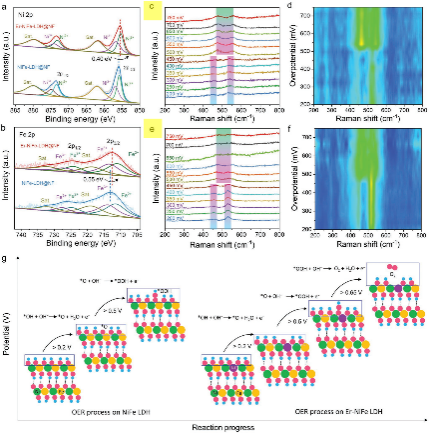
图1 a) Er-NiFe-LDH@NF和NiFe-LDH@NF的Ni 2p和b) Fe 2p XPS谱;OER在0.1 M KOH溶液中的原位拉曼光谱c) NiFe-LDH@NF和e) Er-NiFeLDH@NF;d) NiFe-LDH@NF和f) Er - NiFe - LDH @NF对应的二维原位拉曼光谱等值线图;g)不同偏置电位下NiFe-LDH和Er-NiFe-LDH的OER反应过程的示意图比较
为了深入了解铒掺杂效应的电化学析氧机理,在科学指南针测试机构进行了原位拉曼检测。从200 mV到450 mV的阳极偏置过电位扫描开始,在457和538 cm−1处观察到的两个特征峰可归因于NiFe-LDH@NF表面Ni(OH)2相的Ni-O振动,表明OH -更倾向于吸附在Ni位点上(图5c)。当偏置过电位扫描从450 mV增加到750 mV时,原始表面Ni(OH)2相转变为NiOOH相,其中556和476 cm−1处的两个新特征峰可归因于Ni- OOH振动。形成的NiOOH相通常被认为对OER具有高度活性。然而,NiFe- LDH在450 ~ 750 mV之间呈现NiOOH相的保留,这是由于在高过电位下Ni(OH)2向NiOOH的转变周期缓慢所致(图5d)。而在Er-NiFe-LDH中掺杂Er的作用下,在450 mV之前,随着拉曼强度的增加,OH−在Ni位点的吸附很大程度上促进了OH−在Ni位点的吸附,说明Ni位点的OER活性增强(图5e)。随着偏置过电位在450 ~ 750 mV之间的增大,Ni-OOH的振动模式逐渐变弱,表明Ni-OOH向O2的演化转变(图5f)。简而言之,ER掺杂到NiFe-LDH中,打破了0.5 V后Ni-OOH的原始守恒,促进了*OOH向O2的转化,从而优化了O2释放的缓慢动力学,有利于ER诱导的电子调制(图5g)。
文章摘要:
在Yu Zhu等人的文章中报道了在泡沫镍(Er-NiFe-LDH@NF)上掺杂稀土铒的NiFe-LDH纳米片阵列作为一种高效的OER电催化剂。得到的Er-NiFe-LDH@NF具有优异的OER活性,包括低过电位和活化能,高TOF,以及优越的稳定性,优于NiFe-LDH@NF,商用RuO2和大多数报道的NiFe基电催化剂。
在电化学原位拉曼光谱中,Er掺杂到NiFe-LDH中,Er-NiFe-LDH表现出OH -在Ni位点的有利吸附,并促进Ni-OH转化为Ni- OOH释放O2气体。
理论计算表明,优化后的Er-NiFe-LDH结构由于*O和*OH的键强度平衡,导致反应位点更接近最先进的动力学OER火山的峰值,这与我们的实验观察结果吻合得很好。Er-NiFe-LDH@NF在10 mA cm−2下具有1.43 V的低驱动电位,并且在太阳能驱动的电化学水分解装置中具有较长的驱动周期,显示了其在工业应用中的潜力。
3 原位电化学XRD

文章名称:Hierarchical Architecture Engineering of Branch-Leaf-Shaped Cobalt Phosphosulfide Quantum Dots: Enabling Multi-Dimensional Ion-Transport Channels for High-Efficiency Sodium Storage
期刊信息:1区;IF:32.086;Advanced Materials.
科学指南针测试服务:

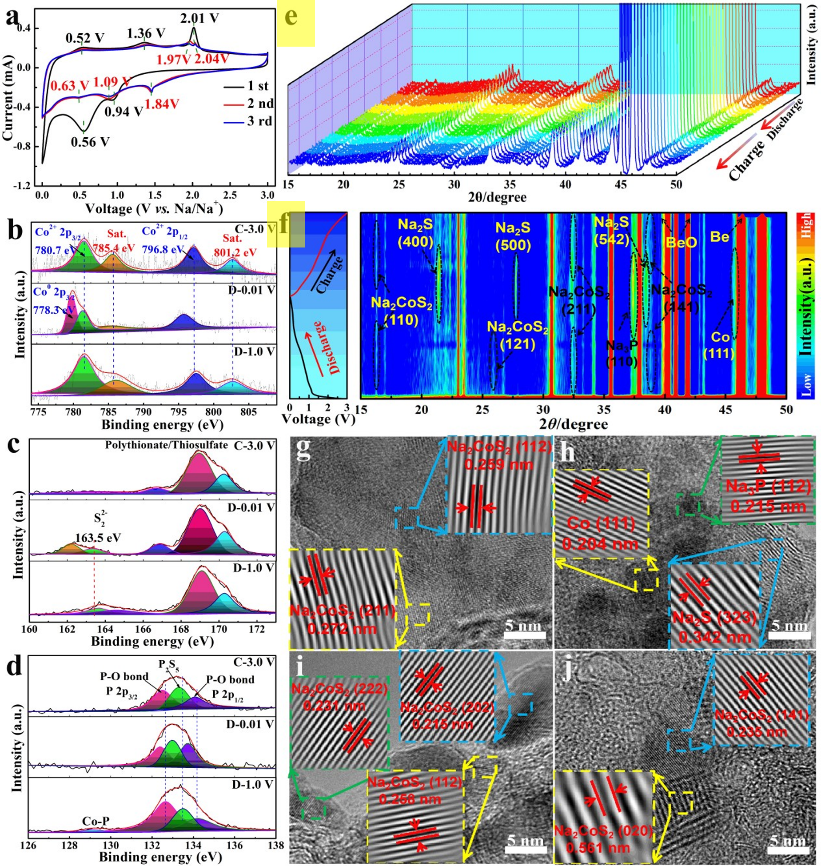
图3 (a) CoPS@C@N-CNF电极在0.5 mV s-1下的CV曲线。CoPS@C@N-CNF电极在不同初始放电和充电状态下的高分辨率XPS光谱:(b) Co 2p光谱;(c) S 2p谱;(d) p2p谱。(e, f) CoPS@C@N-CNF电极初始循环时的原位XRD图谱;CoPS@C@N-CNF电极在不同初始放电和充电状态下的原位HRTEM图像:(g)放电状态为1.0 V;(h)放电状态为0.01 V;(i)充电状态为1.4 V;(j) 3.0 V充电状态。
为了更深入和全面地了解CoPS@C@N-CNF的初始电压依赖性脱钠机理,在第一个循环中进行了原位电化学XRD、非原位HRTEM和SAED测试。从原位XRD图可以看出(图3e, f)。显然,在约37和500的2θ范围内检测到关于BeO和Be的几个反射。在开路电压放电至1.0 V左右时,随着Na+离子的插入,开始了CoPS的第一步插层反应,导致原始CoPS相的衍射峰消失。
同时,中间体Na2CoS2 (JCPDS # 79-2415)在16.00、26.10、33.00、38.30位置出现4个弱峰,匹配的晶面分别为(110)、(121)、(211)、(141)。随着Na+离子不断插入电极,电位下降到0.01 V,即完全酸化状态。清晰地观察到第一次插层反应产生的相消失,同时出现了Na2S (JCPDS # 47-0178)在22.40、27.90和37.70左右的晶面(400)、(500)和(542),以及金属Co在45.9°左右的晶面(111)(JCPDS # 88-2325),有力地提供了Na2CoS2 toNa2S和Co转化反应的证据。在36.10℃左右出现Na3P峰信号(JCPDS # 74-1164),表明P与Na+存在合金化反应。
之后,将电位反向提高到1.4 V左右,Co、Na2S和Na3P峰消失,Na2CoS2重新出现,从而发生了预期的反转转化和脱合金反应。然而,进一步充电至3.0 V时,仍能检测到16.00、33.00和38.30的Na2CoS2峰,这表明在第一次脱氮过程中,CoPS不能完全恢复其原始晶相,这与报道的SIB的CoPSe阳极一致。
值得注意的是,在整个第一个循环中,没有检测到来自磷的特征反射信号,这可能是由于磷的小晶尺寸或无定形特征。更重要的是,CoPS@C@N-CNF在第一个钠化-脱钠循环期间的相变也可以通过异地HRTEM和SAED测量来验证(图S11)。当电位降至1.0 V时,中间产物Na2CoS2完全生成,如图3g HRTEM图像中清晰可见的0.259 nm和0.272 nm的晶格条纹所示,它们分别与(112)和(211)晶体面相关。进一步降低电压至0.01 V(图3h),最终放电产物间距分别为0.204、0.215和0.342 nm,分别分布在金属Co的(220)面、Na3P的(110)面和Na2S的(323)面,表明发生了转化和合金化反应。
当电极连续充电至1.4 V时(图3i),由于Na+的萃取,具有(222)、(202)和(112)面的可逆相变相(Na2CoS2)的晶格条纹再次出现,这可以是维持到3.0 V的带电状态(图3j),进一步表明相演化的不可逆性。这些观察结果与原位XRD结果高度一致,进一步揭示了在持续相变过程中,CoPS与Na+之间发生了多步转化和合金化反应。
文章摘要:
在Wenxi Zhao等人的文章中报道了一种层次化枝叶状CoPS@C@N-CNF结构的合成,该结构采用静电纺丝技术在三维枝状n掺杂碳纳米纤维(N-CNF)上原位生长的叶状金属有机框架衍生的导电碳涂层CoPS量子点,然后进行碳化和同步磷硫化。制备的枝叶状三元复合材料能够综合各组分的优势,包括良好的列弛豫、缩短离子扩散距离、提供充足的电子和电解质、优越的界面电子迁移率、开放的Na+互联通道和充分暴露的活性位点。更重要的是,这是第一次将耦合导电碳的单相CoPS量子点作为SIBs阳极进行精心设计,以彻底探索电化学Na+存储特性。在这些优点的刺激下,CoPS@C@N-CNF具有高容量、持久循环稳定性和良好速率特性的优异Na+存储性能。
4 DEMS

文章名称:A bimetal strategy for suppressing oxygen release of 4.6V high-voltage single-crystal high-nickel cathode
文章链接:https://doi.org/10.1016/j.ensm.2024.103344
期刊信息:1区;IF:18.9;Energy Storage Materials.
科学指南针测试服务:

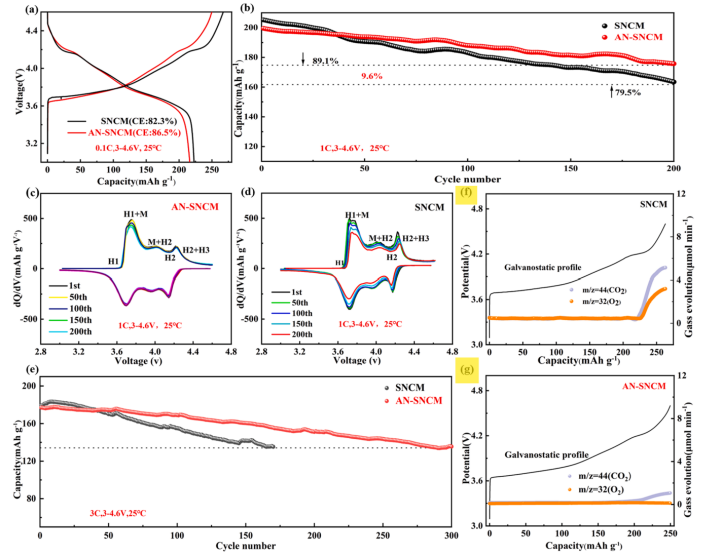
图1 采用SNCM和AN-SNCM阴极的硬币型电池在25℃,3-4.6 v电压范围内的电化学性能(a)原始SNCM和AN-SNCM阴极的初始充放电曲线。(b) 1C电流密度下200次循环的性能曲线。(c) - (d)图2b中两种阴极的dQ/dV光谱。(e)两种阴极在3C电流密度下的循环性能。(f)-(g) SNCM和AN-SNCM电极第二次恒流充电过程中CO2和O2的气体演化曲线。
为了研究SNCM和AN-SNCM在锂去除过程中的氧释放情况,在科学指南针测试机构进行了原位EPR检测,在测试电流密度为20 mAh g−1的情况下,通过DEMS检测其SNCM和AN-SNCM在锂去除过程中的氧释放具体情况如图2f和g所示。SNCM在4.2 V左右表现出O2信号,对应于H2-H3相变过程。随后,CO2信号也会出现,这主要是由于过渡金属与表面有机电解质之间的寄生反应。当然,表面Li2CO3等碳酸盐杂质的分解也会产生少量的CO2。相比之下,在AN-SNCM中检测不到明显的O2信号,在充电结束时仅产生少量的CO2。这为实验中的微结构工程有效抑制晶格氧的释放提供了有力的证据。不同带电状态下O1s的x射线光电子能谱也证实了这一结论。
文章摘要:
高压正极材料,如单晶高镍层状氧化物材料,是实现高能量密度锂离子电池的必要条件,但在使用过程中必须伴随着结构不稳定和晶格氧不可逆释放,存在严重的安全隐患。
为了解决单晶高镍阴极的晶格氧释放问题,我们提出了一种改进的整体阴极策略,通过同步解决界面和晶格不稳定性,确保在高达4.6 V的高压下稳定工作。通过al掺杂,在体相中,我们强化了过渡金属与氧之间的电荷转移,掺杂原子引起的柱状效应有效地缓解了内部应变,从而显著地限制了晶格收缩,从而抑制了氧的释放。另一方面,LiNbO3作为快速离子导体在阴极界面形成的保护层抑制了寄生反应,阻碍了过渡金属溶解引起的晶格氧损失。因此,在4.6 V的截止电压下循环200次后,我们的阴极材料仍然保持89.1%的容量保持率。密度泛函理论(DFT)计算预测了改性阴极材料对氧释放的有效抑制,并通过差分电化学质谱(dem)测试进一步证实了这一点。
本工作为解决单晶高镍阴极在高截止电压条件下的氧释放问题提供了新的思路。
5 原位电化学显微镜

文章名称:Zincophilic Ti3C2Cl2 MXene and anti-corrosive Cu NPs for synergistically regulated deposition of dendrite-free Zn metal anode
期刊信息:1区;IF:10.3;Journal of Materials Science & Technology.
科学指南针测试服务:

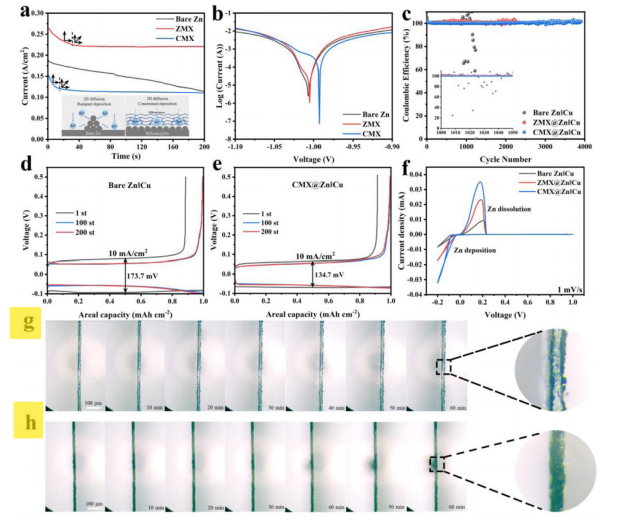
图1 三电极体系中Zn、ZMX@Zn和CMX@Zn电极的电化学性能。(一)Chronoamperograms。(b)线性极化曲线。Zn||Cu、ZMX@Zn| Cu和CMX@Zn| Cu非对称电池的电化学性能(c)固定容量为0.5 mAh cm−2的10ma cm−2 CE轮廓。(d, e)不同周期的电压分布图。(f)扫描速率为1mv s−1时的CV曲线。(g)裸Zn和(h) CMX@Zn电极在5.0 mA cm−2电流密度下的原位光学显微镜图像。
为了进一步证实CMX界面保护层对枝晶生长、化学腐蚀和HER副反应的抑制作用,在科学指南针测试机构进行了原位光学观察。如图1(g)所示,锌沉积10min后,锌表面出现大量不均匀的核。随着电镀时间的延长,锌沉积层不断生长,形成许多突出的部位。特别是电镀60分钟后,暴露的锌表面覆盖着丰富的锌枝晶,表面极不均匀。
相比之下,由于CMX界面保护层的低能垒和有利的亲锌性,锌电极的整体形态演变受到了很好的约束(图1(h))。即使电镀60min后,Zn也趋于均匀沉积在CMX层上,没有出现锌枝晶现象。此外,在辅助资料的视频S1中可以清楚地看到裸Zn的枝晶形成过程。对于疏水CMX@Zn,也可以发现视频S2中CMX表面几乎没有突起和气泡,这表明树突、腐蚀和HER现象得到了有效抑制。结果表明,CMX@Zn阳极在水锌离子电池体系中具有比裸锌阳极更高的稳定性,从而表现出优异的电化学性能。
文章摘要:
锌阳极枝晶生长、化学腐蚀和析氢反应是制约水性锌离子电池(AZIBs)发展的主要障碍。构建界面保护层是缓解阳极副反应的有效途径。采用lewis熔盐蚀刻法制备了具有高亲锌疏水性能的Cu/Ti3C2Cl2 MXene (CMX),并采用简单的自旋涂层构建了CMX界面保护层。CMX涂层可以通过亲锌Ti3C2Cl2 MXene基体提供丰富的成核位点,使电荷分布均匀,从而实现均匀的Zn沉积。
此外,含有抗腐蚀Cu纳米粒子的疏水涂层可以防止Zn阳极受到电解液的腐蚀,有利于抑制化学腐蚀和HER。因此,在0.5 mA·cm -2下,CMX涂层的锌阳极可以实现稳定可逆的镀/剥离,寿命超过1400 h,甚至可以在10 mA·cm -2下,在65 mV下稳定运行700 h。
此外,CMX@Zn在3800次循环中显示出超过100%的高库仑效率,这表明CMX@Zn电极具有优异的锌剥离/电镀稳定性和可逆性。用ZnCoMnO/C (ZCM)阴极组装的完整电池也显示出更高的容量(在0.1 A·g−1时为450.6 mAh·g−1)和循环稳定性(循环1500次后容量保持70%)。这项工作提高了水性锌离子电池(AZIBs)的寿命,并将多功能涂层的研究扩展到其他基于金属阳极的二次电池。









