









 您已经拒绝加入团体
您已经拒绝加入团体

 2025-07-15
2025-07-15
 6474
6474
 0
0
【摘要】 《模拟计算指南》由唯理计算团队编写,提供计算化学的实战方法。本书详细解析轨道定域化技术,如自然键轨道(NBO),用于揭示分子电子结构本质。NBO将正则分子轨道转化为定域化轨道(如BD成键轨道、LP孤对电子),并通过乙醛示例展示其应用。

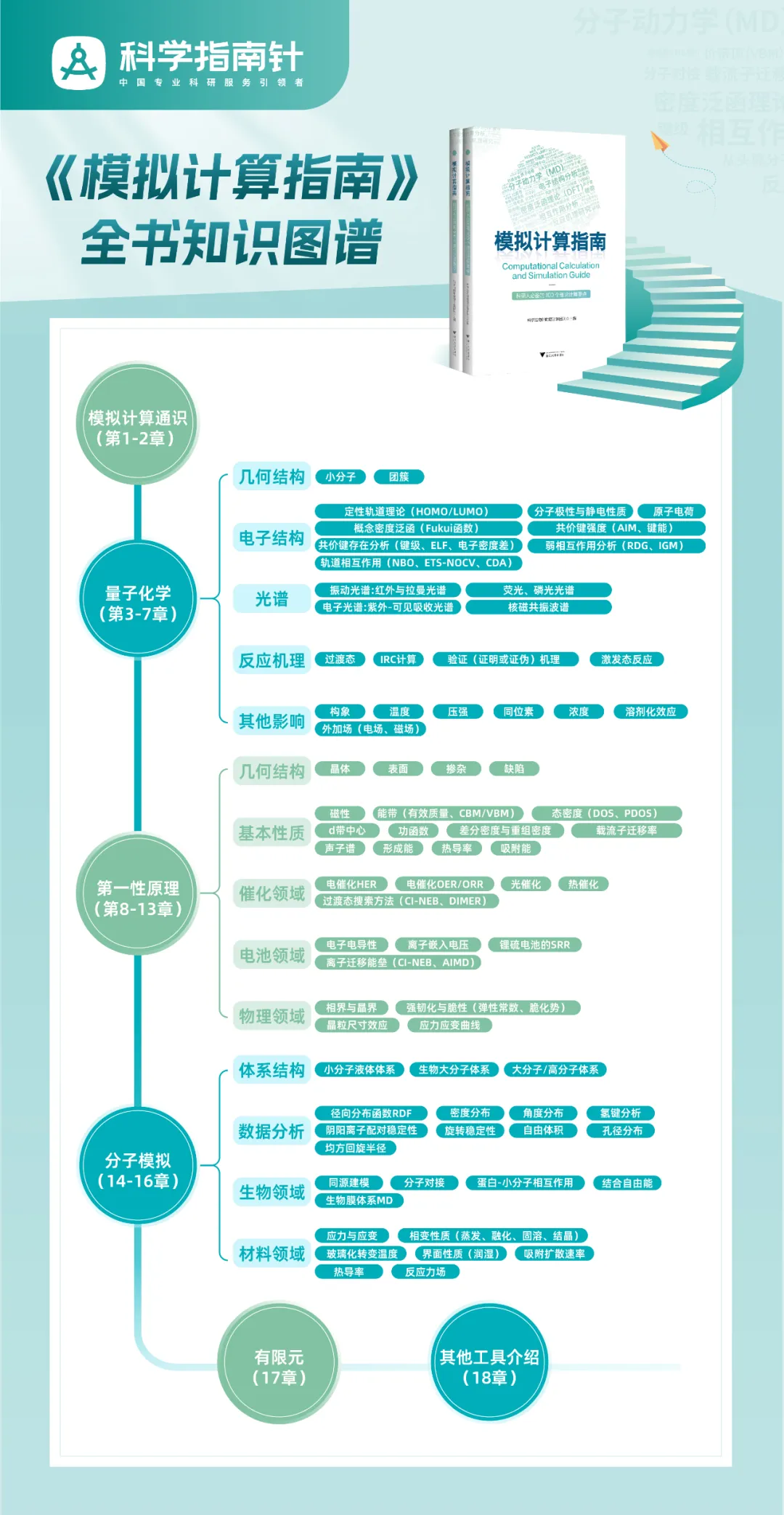
关于本书
《模拟计算指南》是唯理计算工程师团队沉淀7年实战经验、历时一年打造,是一本计算化学快速入门指南、材料模拟计算领域的实用宝典。
“书中详细介绍了从理论计算化学的基本原理到目前国际前沿应用体系的计算模拟思路和方法,有利于读者从多维度理解如何采用理论计算方法来解决复杂科学问题,并帮助初学者从中找到适合自己科研的理论支持和计算解决方案。”
——教育部长江学者、杰青、复旦大学教授
刘智攀
“本书以其实用性和易学性为特色,无论是计算物质科学的初学者还是资深研究者,都能从中获得独特的视角和丰富的知识资源,使其成为该领域内一本极具价值的入门及参考书籍。”
——教育部长江学者特聘教授、华南师范大学教授
赵纪军

↑扫码了解更多书籍及唯理计算信息
01文章介绍
今天我们介绍下《模拟计算指南》的4.1 NBO及其他轨道定域化手段。
在前一章中,我们探讨了许多研究共价与非共价相互作用的手段,由此可以定性或半定量地考察分子中的相互作用。然而在许多情况下,我们不会满足于此,而是希望进一步揭示其中的电子结构本质。例如,通过几何优化,我们能知道两个原子间的键长如何;通过键级计算,又可以知道它们形成了几重键,但我们又会进一步想要知道,其中成键的本质如何,比如有几根σ键、有几根π键、分别通过哪些轨道形成、各自的强度又是多少等。为了解答这些问题,就需要进行进一步的电子结构分析。
轨道定域化是研究轨道相互作用的有力手段。直接计算得到的分子轨道,又叫“正则分子轨道”(canonical orbital,CO或canonical molecular orbital,CMO),属于离域分子轨道,大多数轨道都弥散在整个分子上,而化学家最习惯考虑的轨道图像是定域在原子之间的。轨道定域化就是建立在正则分子轨道与我们更熟悉的基于路易斯结构式的图像之间的桥梁。轨道定域化有多种手段,原理各有不同,其中最知名的莫过于自然键轨道(natural bond orbital,NBO)。
NBO是威斯康星大学的Weinhold等人发展出的一整套波函数分析的方法[20,21],其最主要目的是将分子的电子结构归纳为更接近经验上的路易斯结构式的形式。NBO本身非常复杂,有单独的程序(https://nbo6.chem.wisc.edu/,属于商业软件),其中基础功能被整合在了Gaussian中,我们通常使用Gaussian内置的简化版NBO即可满足大多数需求。
NBO的实现原理颇为复杂,大致可以看作是通过对密度矩阵进行分块对角化而得到一组比较局域化的基函数,再进一步处理得到定域化轨道。NBO轨道被分为4类:内核轨道,记作CR;键轨道,记作BD,相应的有反键轨道BD*;孤对电子,记作LP;里德堡轨道,记作RY。前三者有较强的化学意义。里德堡轨道在字面上指的是某些距离原子核很远、能级很高的高度弥散的轨道,而在NBO的处理过程中,实际上是将所有被NBO程序判断为不属于前三类的轨道统一合并成了里德堡轨道,可以视为轨道分类的“垃圾桶”。对于绝大多数化学问题,我们关心的都是BD、BD*以及LP轨道。
在图4.1中,以M06-2X/def2-TZVP水平下的乙醛为例,使用Gaussian16内置的NBO,展示了几个代表性的NBO轨道。其中,图(a)为占据轨道,图(b)为空轨道。可以明显发现,NBO轨道的特点是高度定域,很容易跟关于化学键的直观感觉对应起来。
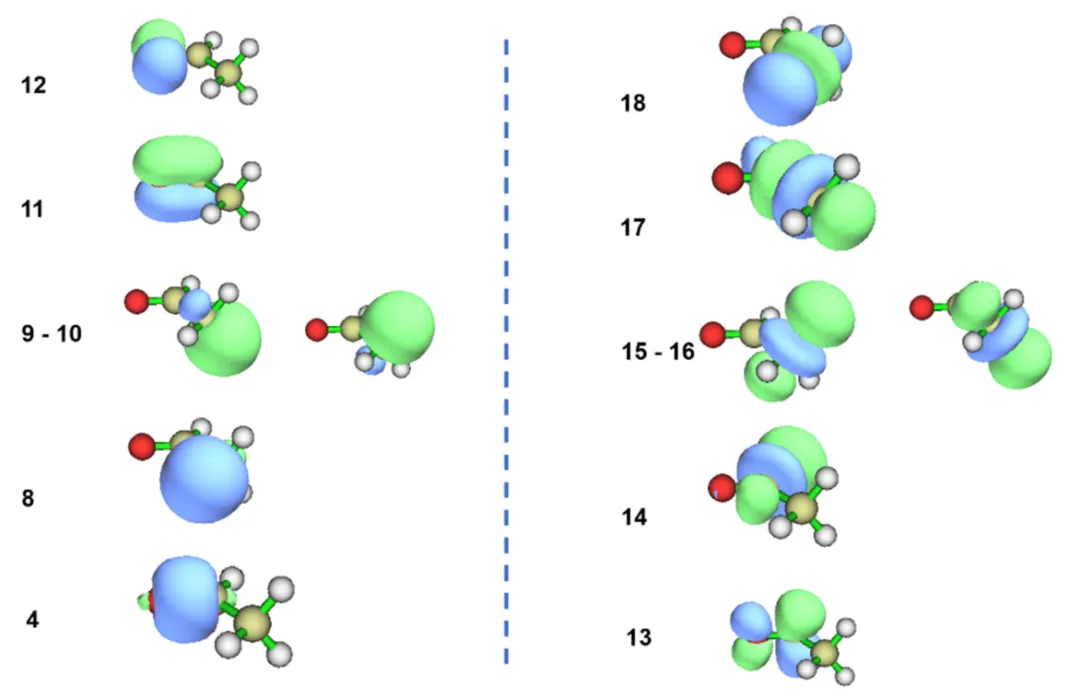
图4.1乙醛的部分NBO轨道等值面
图4.1中,轨道8~10为C—H键的成键轨道,被称为BD(C—H);轨道11为BD(C一O),而轨道12对应氧上的孤对电子,属于LP(O)。从轨道13开始为反键轨道,分别是BD*(C—O),共计4个BD*(C—H)和1个BD*(C—C)。轨道形状也非常直观地与我们关于化学键的想象契合。通过观察NBO轨道,可以容易地知道,乙醛中的羰基C一O由一根σ键和一根π键构成,两者对应的成键轨道分别为轨道4和轨道11,分别填充2个电子,而反键轨道都是空的,从而形成一根双键。
识别出定域化轨道的存在只是NBO分析的第一步,在此基础上还可以得到许多其他信息。例如,乙醛里的羰基碳原子上连接了3个不同的基团,必然发生不等性杂化,那么这个碳原子与每个基团成键时使用的分别是什么杂化轨道?以上述结构为例,在输出文件中,可以找到如图4.2所示段落,描述了每个NBO轨道由哪些原子轨道组成。
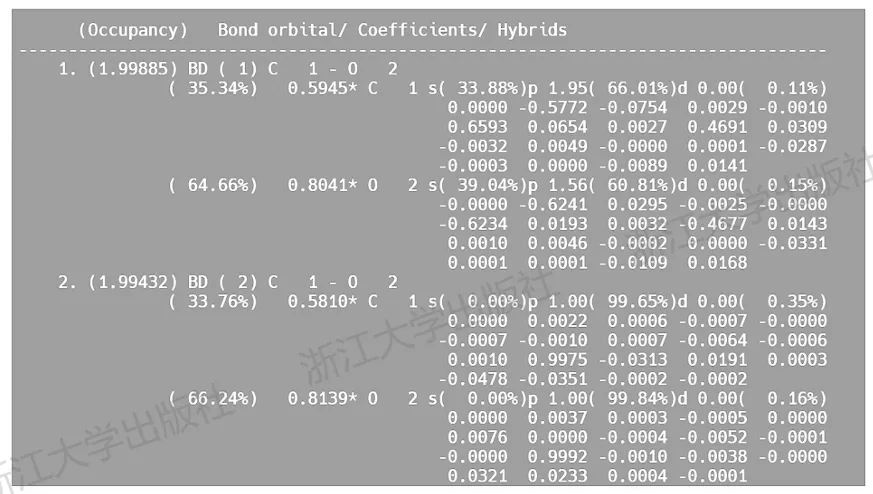
图4.2乙醛的C—0NBO轨道成分
其中第1和第2个轨道(值得注意的是,此处显示的顺序与能量顺序并不相同,要加以甄别)分别是羰基碳氧键的σ和π成键轨道。在轨道1中,羰基碳原子采取sp1.95杂化轨道与氧原子的sp1.56杂化轨道组合形成σ键。在轨道2中,羰基碳原子贡献了33.76%,利用纯p轨道与氧原子的p轨道形成π键。类似地,可以在后续输出文件中发现,这个羰基碳原子利用sp2.20轨道与氢原子成键,又利用sp1.83轨道与甲基成键。
羰基C—O键具备极性,其成键轨道向电负性较强的原子倾斜,反键轨道则主要由电负性较弱的原子贡献,通过原子贡献的分析,也容易验证这一点。图4.2中,在轨道1中,碳原子贡献了35.34%,而对应的反键轨道中,碳原子则贡献了64.66%。通过观察轨道形状也可以直观看出,碳氧键的BD*轨道中碳原子贡献的轨道瓣明显更大。
除了轨道及轨道成分外,NBO还可以分析轨道之间的二阶相互作用,这是研究超共轭的利器。事实上,物理有机化学中关于超共轭的观念,很大程度上就是依赖于以Houk等人为代表的研究者借助NBO的工具提炼出的结论。仍然以乙醛为例,我们知道乙醛中存在多组超共轭作用,其中比较重要的是α-C—H对π*(C—O)的超共轭,以及氧原子孤对电子对σ*(C—H)的超共轭,如图4.3所示。
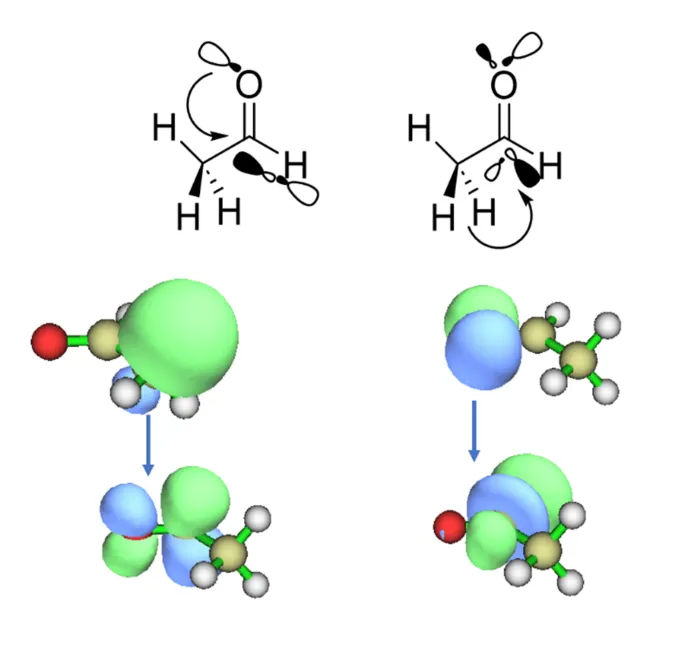
图4.3乙醛分子中存在的部分超共轭作用
通过对NBO轨道的可视化,容易看到参与超共轭的轨道在空间上的反向共平面关系;在输出文件中,E(2)部分记录了相应的超共轭作用的能量贡献,如图4.4所示。在这里记录了庞大的超共轭作用的列表,从中识别出感兴趣的轨道,可以发现上述绘制出的LP(O)→BD*(C—H)和BD(C—H)→BD*(C—O)两组超共轭分别贡献了1.00kcal/mol和6.23kcal/mol的稳定化能。图4.4中的最后两列,E(j)-E(i)以及F(i,j)则分别对应参与超共轭的两个轨道的能量差和空间重叠程度,F的数值越大,意味着空间重叠越好。
关于超共轭能量的定量对比是研究许多化学问题的重要工具,哈佛大学的Kwan在其序号为Chem106的讲义中(https://ekwan.github.io/notes.html#experimental-organic-chemistry),广泛运用NBO手段定量讨论各种相互作用对化学现象的影响,非常值得一读。
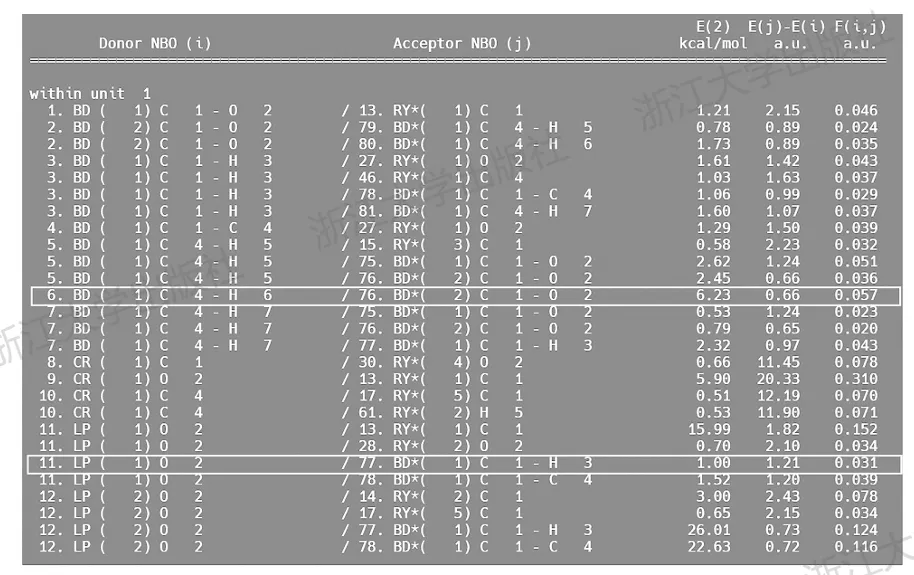
图4.4Gaussian输出文件中与乙醛分子超共轭相关的部分
除NBO外,还有许多其他轨道定域化手段。Multiwfn程序支持基于主流的量子化学程序输出的波函数文件进行轨道定域化,支持Pipek-Mezey和Foster-Boys两种方法。它们的原理与NBO不同,是基于对正则分子轨道的直接线性组合,不会将轨道进行分类,也无法直接得到超共轭能量等数值。
与NBO相比,它们的优点有两个:一是方便,不需要调用NBO程序,基于任何波函数文件即可进行;二是普适性较强。NBO方法存在的最主要局限在于其对于轨道进行分类、搜索的算法相当复杂,对于许多电子结构怪异或电子离域性很强(如新奇的过渡金属配合物、不能用通常路易斯结构式描述的新颖化合物等)的场合并不适用,而恰恰正是在这种情况下人们更需要通过定域化轨道的方法窥探成键的本质。这种情况下,基于Pipek-Mezey或Foster-Boys方法的轨道定域化要普适得多。至于轨道的形状,这些算法得到的结果与NBO轨道类似,都呈现出与我们对于化学键经典认识相契合的成键、反键轨道或孤对电子的形状,不必单独阐述。
在Catal.Sci.Technol.,2022,12:880-893中,作者就通过Pipek-Mezey方法对Os(VⅢ)促进的烯烃双羟基化反应过程中Os—O键的演变情况进行了深入研究,阐明了其中Os—0多重键中每一重共价作用的轨道本质以及在反应过程中的变化规律。

多位专家力荐 超全实战指南


↑扫码了解更多书籍及唯理计算信息








