









 您已经拒绝加入团体
您已经拒绝加入团体

 2026-05-20
2026-05-20
 17
17
 0
0
【摘要】 围绕 Li-Si-C 体系 NEP 机器学习势函数、DFT、AIMD、主动学习和数十万原子级模拟展开,突出接近 DFT 精度与约 7 万倍加速。

科学指南针唯理计算工程师廖宇杰的机器学习势函数研究成果,已发表于《ACS Applied Materials & Interfaces》。该研究针对 Li-Si-C 三元体系搭建 NEP 模型,在模拟效果接近 DFT 计算精度的前提下,实现相较传统 AIMD 模拟约 7 万倍运算提速,同时可稳定开展数十万原子级模拟工作,为多元材料体系计算研究提供可靠技术支撑。

NEP 机器学习势函数是什么?
NEP 机器学习势函数是依托大数据学习搭建的新型材料计算模型,它主要通过学习提炼 DFT 计算得出的能量、原子作用力等核心数据信息,简化后续分子动力学模拟的运算流程。该模型能够在维持稳定模拟精度的同时,有效提升大规模原子体系的计算运行效率,相较于传统经验势函数,更适配成分复杂、作用环境多样的多组分材料体系研究。
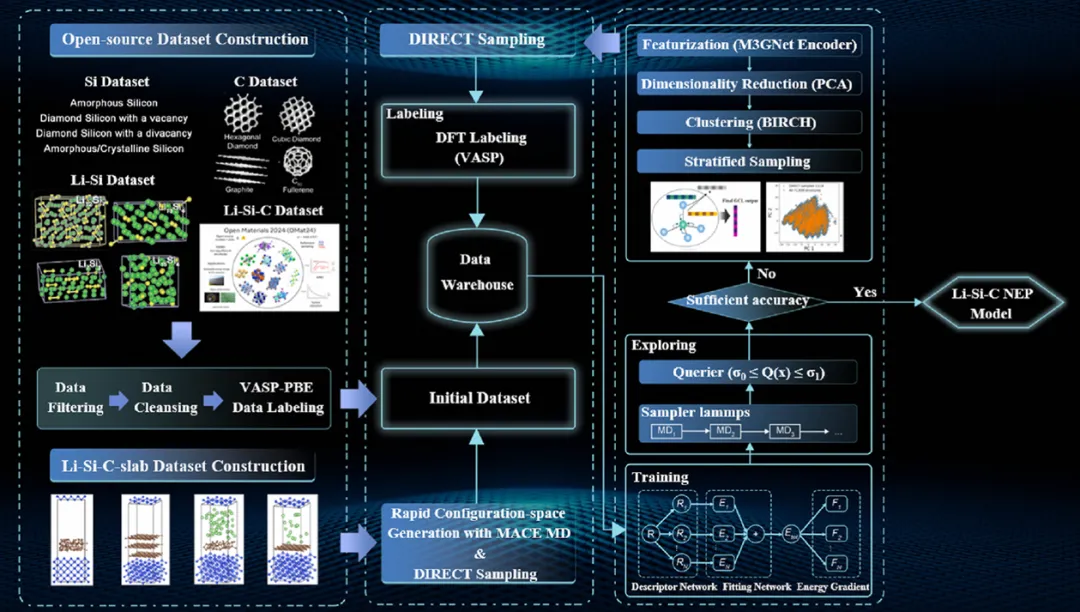
用于设计 Li−Si−C NEP 力场模型的示意工作流程
为什么 Li-Si-C 体系需要机器学习势函数?
传统材料计算手段存在明显应用局限,这也是 Li-Si-C 三元体系研究引入新型势函数的核心原因。DFT 计算结果精准,但整体运算成本较高;AIMD 模拟适合分析原子动力学变化,面对复杂界面与大尺度原子集群时运算周期过长。Li-Si-C 属于多组分复合体系,传统方法难以兼顾精度与效率,引入 NEP 模型能够很好补齐这一研究短板。
主动学习如何提升 NEP 模型建模效率?
主动学习是优化 NEP 模型搭建效率与覆盖范围的重要手段,该方法可以智能筛选出代表性更强的结构样本进行训练。依托这一优化方式,本次研究仅选用原始数据池内 4.3% 的结构样本,就能够完整覆盖体系 90% 以上的特征空间,既减少了前期数据采集与整理的工作量,也能通过持续参数迭代,不断拉近模拟结果与 DFT 理论数值的差距。
7 万倍加速对材料计算有什么价值?
实现约 7 万倍的 AIMD 模拟提速,能够大幅缩短各类复杂材料体系的科研测算周期。搭配模型具备的数十万原子级模拟能力,科研人员可快速完成大尺度材料微观机理探究、材料基础性质预测等常规研究工作,减少算力与时间成本消耗,也为后续 AI + 材料研发相关科研训练积累了成熟落地案例。
常见问题 FAQ
NEP 机器学习势函数是什么?
NEP 即机器学习势函数,是面向材料模拟研发的智能计算模型,以 DFT 标准数据为学习基础,简化分子动力学模拟流程,兼顾运算效率与模拟准确度,可适配 Li-Si-C 等多元体系,区别于通用性较弱的传统经验势函数。
Li-Si-C 体系为什么适合用机器学习势函数?
Li-Si-C 三元体系组分复杂、微观界面结构繁多,DFT 与 AIMD 组合计算耗时久、算力消耗大。NEP 模型可适配其复杂化学作用环境,依托高效运算优势,轻松完成数十万原子尺度的模拟工作,契合该体系基础机理研究需求。
主动学习在 NEP 模型中有什么作用?
主动学习能够精准筛选优质训练样本,大幅降低 NEP 模型训练所需的数据体量,仅用少量结构数据即可覆盖绝大多数体系特征。同时可动态优化模型参数,持续优化模拟精度,是实现小数据建模、高效运算的核心辅助方式。
7 万倍 AIMD 加速是否会影响模拟精度?
本次优化提速并未牺牲核心模拟精度,NEP 模型运行得出的结果整体接近 DFT 计算精度。提速主要通过简化冗余运算流程实现,仅优化运算速率,不会改变材料能量、结构演化等核心模拟逻辑,可满足常规科研数据使用标准。








