









 您已经拒绝加入团体
您已经拒绝加入团体

 2021-11-16
2021-11-16
 3656
3656
 0
0
【摘要】 锂离子电池由于其高能量密度/效率和长循环寿命,已经征服了电动汽车、便携式电子产品和机器人的重要市场。

文章背景
锂离子电池由于其高能量密度/效率和长循环寿命,已经征服了电动汽车、便携式电子产品和机器人的重要市场。
对更高能量密度(如4400Wh·kg-1)需求的显著增长促使我们探索传统LiB之外更具侵略性的化学物质,包括高电压容量阴极和转化型阳极。
然而,这种化学反应通常伴随着高电化学反应性和不稳定的电极-电解质界面,使得保持令人满意的循环寿命具有挑战性。
因此,形成稳定的电致发光元件以减轻反应性电极和电解质的降解至关重要。
成果简介
麻省理工李巨教授课题组证明了一种合理设计的磺酰胺基电解质可以通过稳定锂金属负极 (LMA) 和高压 LCO 正极上的电极 - 电解质界面 (EEIs) 来大大提高高达 4.7 VLi 的高压循环稳定性 .
使用磺酰胺基电解液,商用 LCO 正极在 4.55 VLi 和 4.6 VLi 的高充电电压下分别在 200 和 100 次循环后保持其容量的 89% 和 85%,显着优于传统的碳酸盐基电解液。
表面降解、阻抗增长和气体逸出和 Co 溶解方面的有害副反应得到了很好的抑制。这项工作展示了一种设计新电解质以实现高能 Li||LCO 电池的有前景的策略。
图文导读
采用密度泛函理论(DFT)计算来比较 EC/DMCF3SA 分子与高度脱锂的 LCO 表面之间的氧化反应能量学(图 1a 和 b),发现DMCF3SA分子具有更高的抗氧化性。
图 1c 显示,磺酰胺基电解液的氧化开始为 ~5 VLi,没有对应于 Al 腐蚀的峰,而碳酸盐基电解液的氧化开始于 ~4.2 VLi。
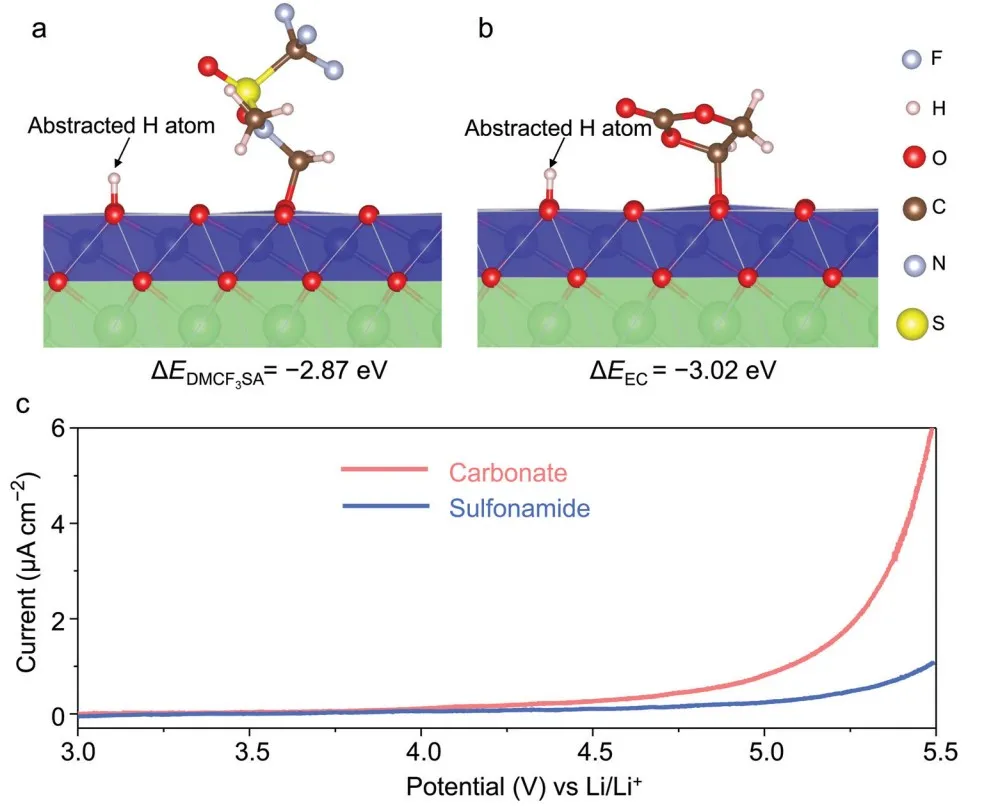
图1
使用磺胺基电解质的 Li||A-LCO 电池的 4.55 VLi 循环性能得到显着改善。该电池的初始容量为 200.8 mAh g-1,在 200 次循环后分别保留了 89% 和 99.74% 的容量和操作中电压(图 2a 和 c)。
~99.84% 的优异平均 CE(图 2b)表明 A-LCO 阴极和电解质之间不希望的副反应在很大程度上得到抑制。评估了 4.55 VLi 的倍率性能(图 2d-f),使用磺胺基电解质的 Li||A-LCO 电池表现出更高的可逆容量。
此外,还在 4.6-4.7 VLi 的更高充电电压下评估了另一种商业 T-LCO 阴极。尽管具有两种电解质的 Li||T-LCO 电池达到了类似的高初始容量~220 mAh g-1,但容量保持率显示出显着差异(85% 对 57%,图 2g 和 h)。
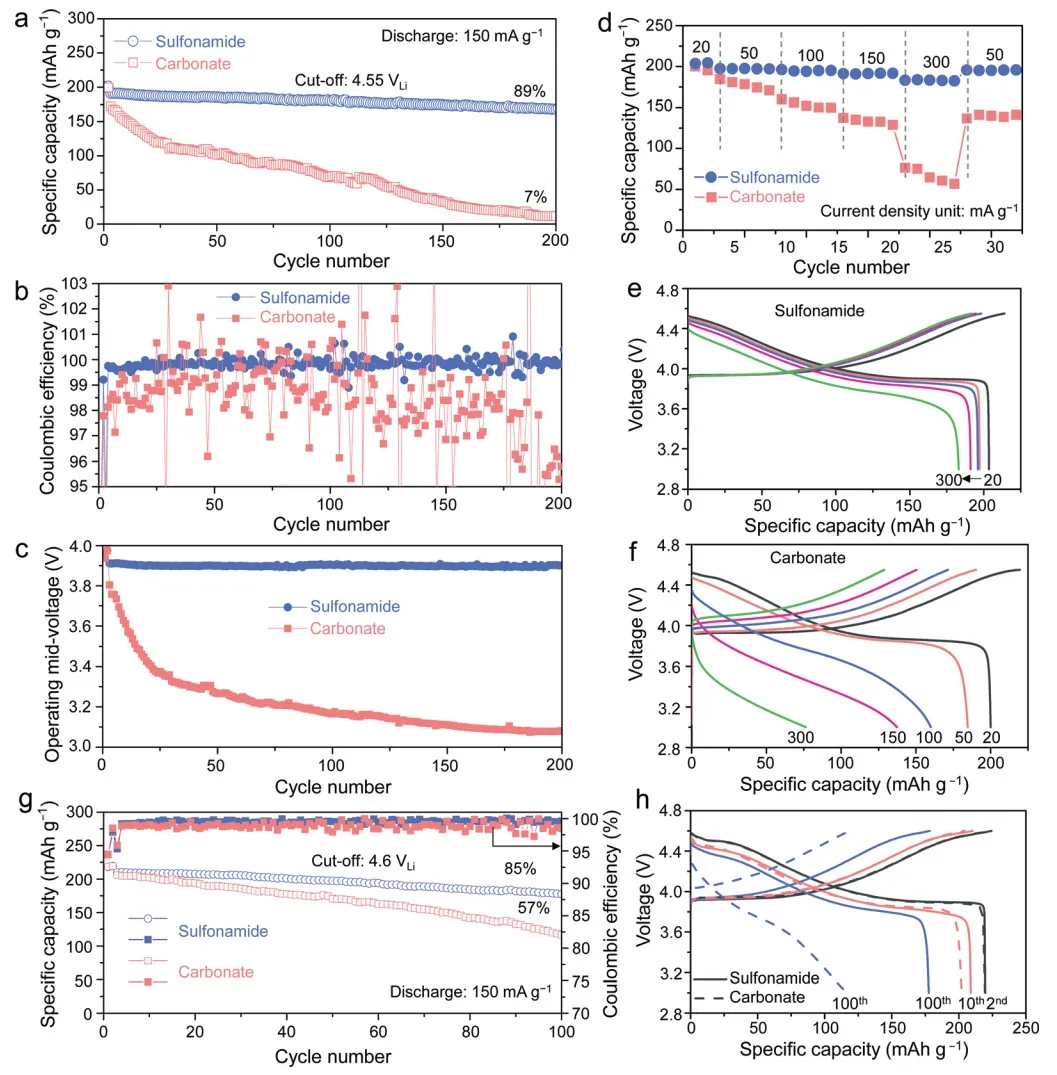
图2
为了阐明降解机制,研究集中在不同电解质中以 4.55 VLi 循环后的 A-LCO 阴极。首先对 Li||A-LCO 电池进行恒电流间歇滴定技术 (GITT),以测量循环后的阻抗增长(图 3a 和 b)。
对于参比电解质,在 200 次循环后观察到严重的阻抗增长和巨大的过电位(图 3b,平均过电位为 758±80 mV)。相比之下,对于EC/DMCF3SA电解质,200 次循环后的过电位小一个数量级(图 3b,平均过电位为 15±4 mV),这与上面报道的更好的容量保持率和倍率性能一致。
在将循环的 A-LCO 阴极充电至 4.55 VLi 后,在使用EC/DMCF3SA电解质(图 3c)循环的阴极中注意到比使用碳酸盐基电解质(图 3d)更高且更均匀的 Co 价。
几个代表性粒子的白线分布的绘制直方图进一步证实了这一结果(图 3e)。如图 3f 和 g 中的黄色箭头突出显示,在碳酸盐基电解质中循环的 A-LCO 在大颗粒中显示出明显的裂纹,而在磺酰胺基电解质中循环的 A-LCO 保持完整(图 3h 和 i)。
这一结果表明,通常在多晶 NMC 阴极中观察到的 SCC 行为也存在于具有单晶微观结构的深循环 LCO 阴极中,通常认为其不易开裂。虽然在碳酸盐基电解质中循环的 A-LCO 的电阻层厚约 11 nm(图 3j),但它被磺酰胺基电解质抑制(约 5 nm 厚,图 3k)。
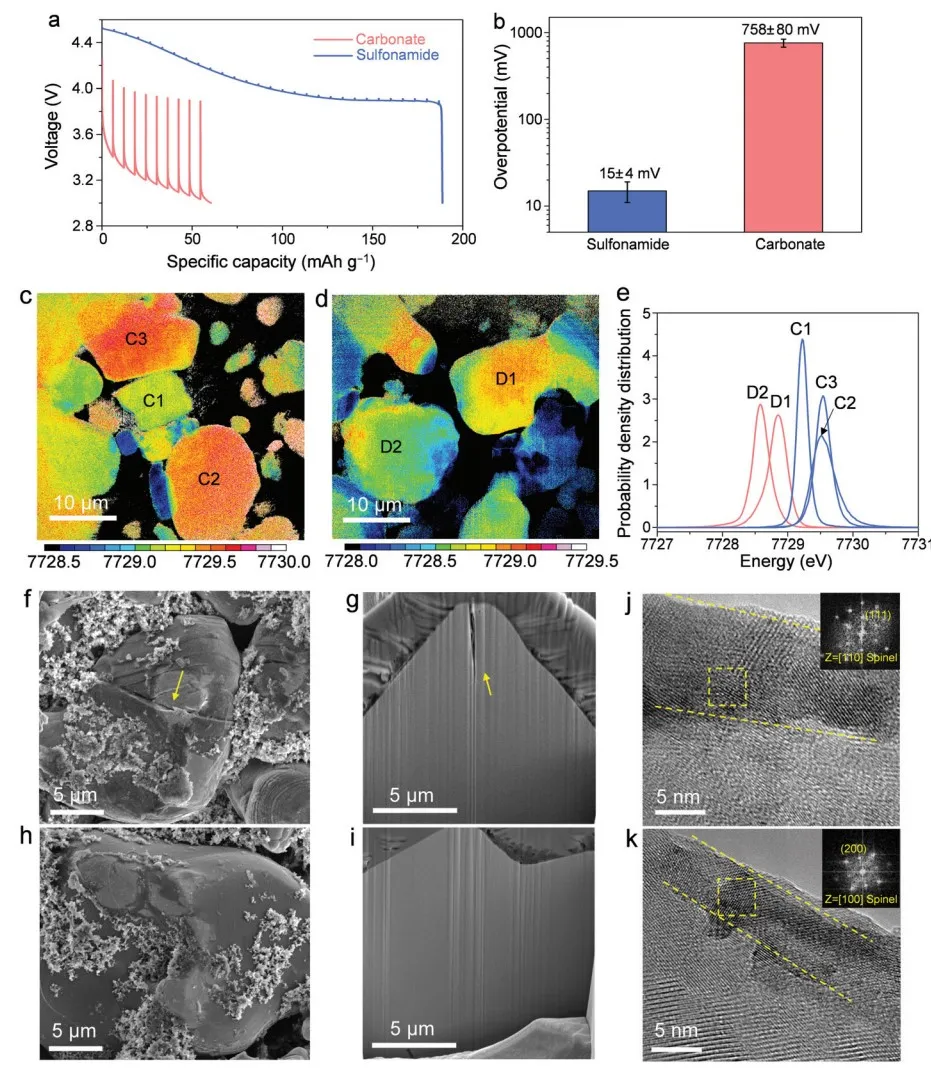
图3
进行差示电化学质谱 (DEMS) 以监测充电至 4.7 VLi 期间的气体逸出。当 A-LCO 在碳酸盐基电解质中充电至 4.5 VLi 以上时,明显的 CO2 气体开始释放(图 4a),证实了电解质组分的氧化。
相比之下,磺胺基电解液没有检测到明显的气态产物,包括 CO2、O2(图 4b)或其他可能的气体(图 S12),表明其具有出色的抗氧化性。在将 LCO 从开路电位 (OCP) 充电到 4.8 VLi(图 4c)时,观察到从 3.9 VLi 到 4.2 VLi(图 4d)的红外光谱的明显变化。
注意到 SQO 区域~1260 cm-1 处的峰值,最早出现在~4.0 VLi。这可能归因于 LiFSI 或 DMCF3SA 的分解,因为它们都具有 SQO 基团。
从 4.2 VLi 到 4.8 VLi 进一步充电后,光谱几乎保持不变(图 4d),表明 CEI 对氧化具有良好的化学稳定性。充电至~4.4 VLi 时,LCO 表面上的溶剂氧化通过 C 1s 的 XPS 分析(图 4e)证实,因为高结合能(即对应于氧化碳,如 C-O 和 C-SOx)的峰值强度增加从 4.2 VLi到 4.4 VLi,表明由 DMCF3SA 分解形成的表面物质的生长。
从 4.4 VLi和以上的 F 1s 光谱(图 4f)注意到 S-F 物种,这可能归因于具有 S-F 键的 LiFSI 盐的分解,而不是具有 CF3 键的 DMCF3SA 溶剂。
C-F 峰强度高于 S-F 峰强度(图 4f)意味着 LCO 表面上的 CEI 主要来自 DMCF3SA 溶剂的分解,而不是 LiFSI 盐。
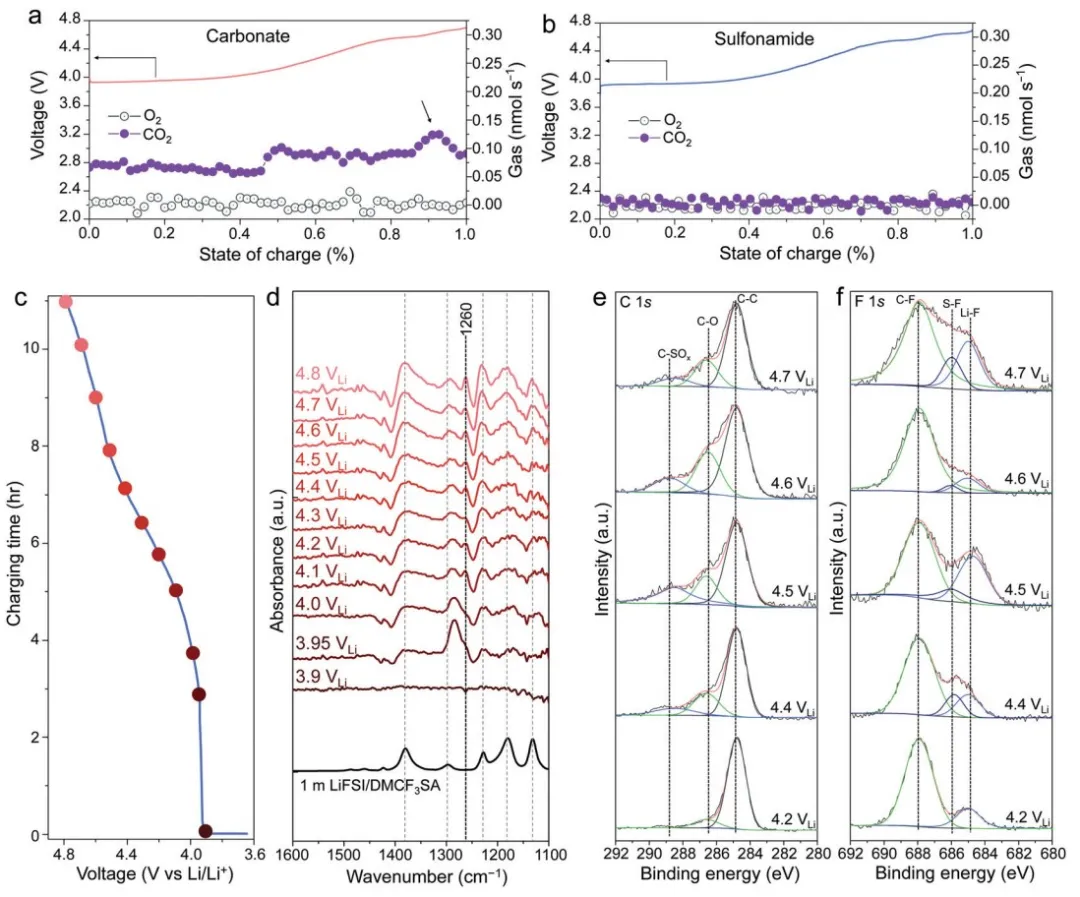
图4
采用了一种行之有效的 Li||Li 配置方法来评估锂基体上的锂金属剥离/镀层 CE(图 5a)。
与碳酸盐基电解液 (CE~65.7%) 相比,我们的磺酰胺基电解液在镀锂和脱锂过程中表现出更高的 CE~99.7% 和更小的过电位。在基于碳酸盐的电解液中循环的 LMA 上可以看到枝晶状锂沉积物(图 5b)。
这种高表面积形态可以促进 LMA 和电解质之间的有害副反应,从而导致 LMA 可逆性较差。此外,检测到来自电子色散谱(EDS)光谱的明显 Co 信号(图 5c) ,它被认为是从 LCO 阴极迁移的。对于基于磺酰胺的电解质,锂沉积物的形态往往是表面积较小的大颗粒(图 5d),而没有交叉 Co 的存在(图 5e)。
图 5f 中较低的 C 1s 强度表明磺酰胺的溶剂分解少于碳酸酯。F 1s XPS 光谱(图 5g)显示磺胺基电解液中形成的 SEI 中的 LiF 含量高于碳酸盐基电解液中形成的 LiF 含量,这可以有效地稳定 SEI。
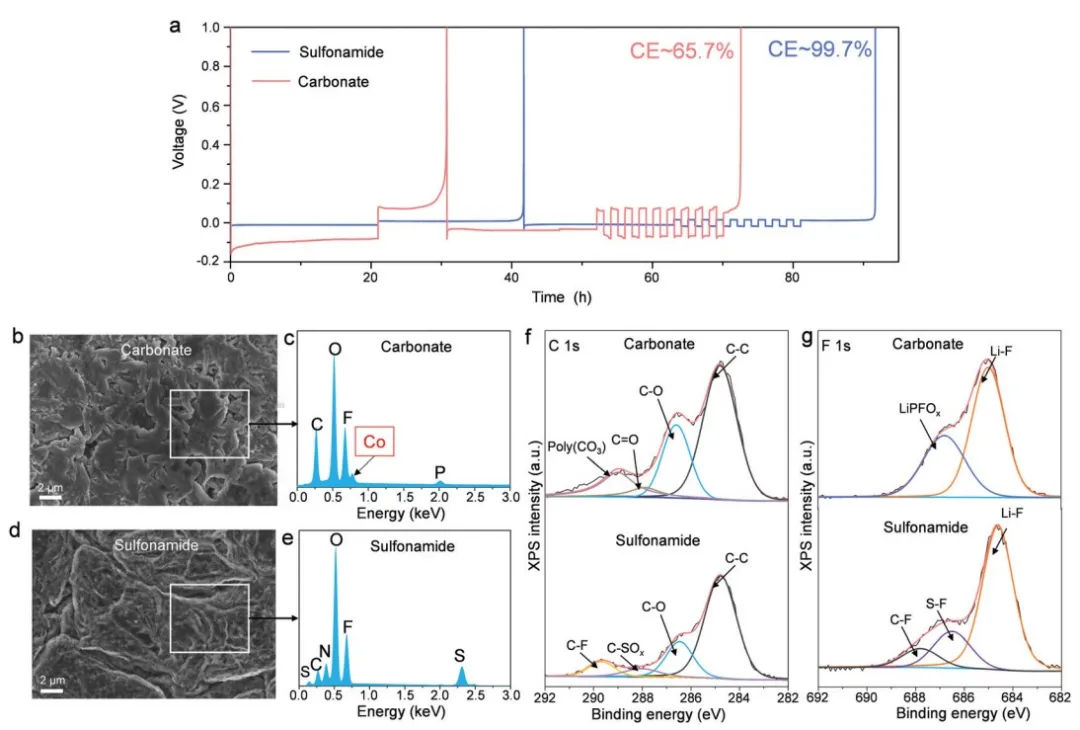
图5
总结与展望
这项工作设计并展示了一种磺酰胺基电解质,以稳定高压锂金属||LCO 电池中的电极-电解质界面。
基于磺酰胺的电解质成功地实现了商业 LCO 正极的优异循环性能,在 4.55 VLi 和 4.6 VLi 的高充电电压下,分别在 200 和 100 次循环后具有 89% 和 85% 的高容量保持率。
磺酰胺基电解质通过抑制表面降解、阻抗增长以及气体逸出和 Co 溶解方面的副反应,有效地稳定了阴极 - 电解质界面。
除了正极之外,磺酰胺基电解质还与锂金属负极具有优异的相容性,由于形成了有利的沉积形态和稳定的 SEI,锂的剥离/镀层 CE~99.7%。
我们的工作提出了一种设计新电解质以实现高电压和高能量锂金属||LCO 电池的有前景的策略。
文献链接
https://pubs.rsc.org/en/content/articlelanding/2021/ee/d1ee01265g#!divRelatedContent
公众号测试万事屋后台回复【获取原文1112】可获取文献全文









