









 您已经拒绝加入团体
您已经拒绝加入团体

 2025-11-11
2025-11-11
 3974
3974
 0
0
【摘要】 本文解读了《模拟计算指南》一书中关于过渡态与IRC(内禀反应坐标)的章节,系统介绍了过渡态的定义、寻找方法(如Berny算法)、虚频验证、IRC计算流程、自由能面分析以及表观能垒等关键概念。内容结合实例(如S_N2反应)阐述计算化学中的反应机理研究,旨在帮助科研人员深入理解模拟计算在反应动力学中的应用。

关于本书
《模拟计算指南》是唯理计算工程师团队沉淀7年实战经验、历时一年打造,是一本计算化学快速入门指南、材料模拟计算领域的实用宝典。
“书中详细介绍了从理论计算化学的基本原理到目前国际前沿应用体系的计算模拟思路和方法,有利于读者从多维度理解如何采用理论计算方法来解决复杂科学问题,并帮助初学者从中找到适合自己科研的理论支持和计算解决方案。”
——教育部长江学者、杰青、复旦大学教授
刘智攀
“本书以其实用性和易学性为特色,无论是计算物质科学的初学者还是资深研究者,都能从中获得独特的视角和丰富的知识资源,使其成为该领域内一本极具价值的入门及参考书籍。”
——教育部长江学者特聘教授、华南师范大学教授
赵纪军

↑扫码了解更多书籍及唯理计算信息
01文章介绍
今天我们介绍下《模拟计算指南》的6.1过渡态与IRC。
在第1章中,已经介绍了化学反应的速率理论。通过对反应过程中的中间体和过渡态进行寻找,即可得到各基元反应的热力学和动力学信息,进而研究反应机理。
在势能面上,过渡态是沿着反应坐标方向的能量极大值点,而在垂直于反应坐标的各方向上都是能量极小值点,即势能面上的一阶鞍点。在量子化学程序中,想要寻找过渡态,最普遍的方法是Berny算法。在这个算法中,需要输入足够接近过渡态的初始结构,随后程序会试图寻找与其结构接近的某个过渡态。过渡态的收敛范围通常比基态(在本节中,“基态”指的是势能面上的极小值点)窄得多,这意味着只有当初始构型与真正的过渡态非常接近时,才能成功收敛到所寻找的过渡态结构。这既要求计算化学工作者对物质结构和反应性有深入的理解,其工作量也比基态结构优化大出不少。即使是经验非常丰富的研究者,在寻找过渡态时也时常碰到难以收敛到感兴趣的过渡态的情况,往往需要大量尝试和艰苦的努力。
一个结构是过渡态的必要条件是有且只有一个虚频,因此进行过渡态搜索后总要进行频率计算,以验证是否正确找到了过渡态。虚频的方向对应了过渡态作为能量极大值点的方向,即与反应坐标相同。在大多数情况下,通过观察虚频振动模式,即可判断过渡态是否对应了所感兴趣的反应。
借助在上一章中介绍过的方法,可以使用Gauss View方便地查看虚频振动模式。以氟离子与氯甲烷的SN2反应过渡态为例,频率计算得到一个显示为-637.68cm⁻¹的振动模式,是该结构的唯一虚频。观察该振动模式的动画(如图6.1所示,振动模式以箭头方式表现),可知该虚频模式中碳原子在F和Cl之间转移,从而可以判断该过渡态对应于SN2反应。需要注意的是,虽然Gauss View将虚频的振动频率显示为-637.68cm⁻¹,但这一表示方法的实际含义为637.68icm⁻¹,在报道虚频频率时务必写作包含虚数单位的形式。
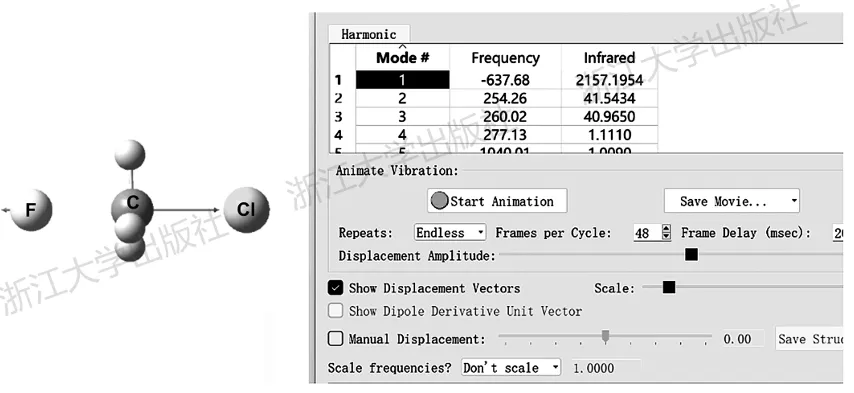
图6.1氟离子与氯甲烷的SN2反应过渡态及其虚频
过渡态虚频的大小和红外强度的含义与普通的红外光谱相同。其数值大小反映了该振动模式的力常数和有效质量,而红外强度反映了该振动模式中偶极矩的变化。因此,以氢原子转移为代表的反应中,有效质量很小,虚频振动频率数值很大,可以达到几千个cm⁻¹;而诸如骨架构象旋转、重原子转移等有效质量很大的反应,其过渡态虚频则很小,可能只有几十个cm⁻¹。与此同时,力常数则反映了势能面对于几何结构的二阶导数,因此当过渡态处势能面非常平缓时,力常数很小,相应地,虚频频率也就很低;而如果过渡态处能量变化尖锐,则虚频频率也就较高。
过渡态与基态一样,可以进行各种电子结构分析。通过标准的频率计算和单点计算,可以得到过渡态的自由能,进而用于求算反应能垒。
通过观察虚频振动模式,可以判断过渡态是否对应于所感兴趣的反应。而很多情况下,我们还希望知道在过渡态所对应的反应过程中几何结构沿着反应坐标是如何连续变化的,这就需要IRC(intrinsic reaction coordinate)计算。
在IRC计算中,从优化好的过渡态结构出发,在与过渡态几何优化相同的计算水平下,程序尝试寻找势能面上通过过渡态的能量最低路径,即为该过渡态所连接的反应路径。在反应路径上,从过渡态结构出发不断走步,能量不断下降,当能量趋于不变或上升时,即认为达到了过渡态所通往的极小值点。通过这种方法,可以知道过渡态是连接哪两个极小值点的,也可以知道在整个反应坐标上结构、能量及其他各种性质是如何变化的。
IRC计算的时间成本比构型优化多得多。这是由于为了得到准确的IRC路径,需要每隔特定步数就通过频率计算得到当前点上的精确力常数。为了得到完整的IRC路径往往需要走几十上百步,可能会进行几十次频率计算,耗时相当高。因此,如果只是希望知道过渡态是否找对,通常通过虚频振动模式判断即可;即使要进行IRC计算,通常也只需走到能看出所连接的极小值点的大致形状即可,不必追求把反应曲线跑完。只有当虚频振动模式比较模糊、令人难以下定论,或者希望对反应过程中几何和电子结构的变化进行更加深入的研究时,才会进行IRC计算。
图6.2中展示了上文提到的SN2反应过渡态的IRC能量曲线。
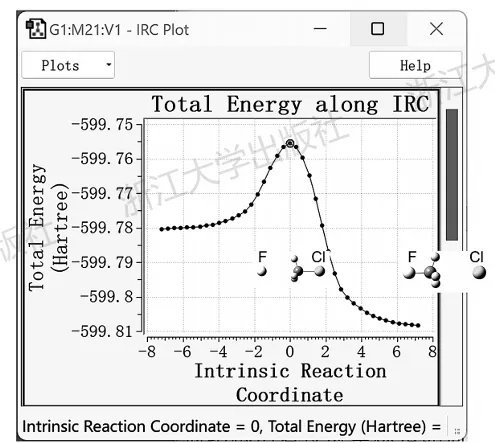
图6.2氟离子与氯甲烷的SN2反应过渡态的IRC及其两端几何结构
其中过渡态被定为原点,左右方向任意,分别对应其连接的两个结构,即两种卤代烃与卤离子的复合物。随着反应的进行,能量曲线呈现出平滑的先升后降趋势。需要注意的是,量子化学程序进行构型优化、IRC计算等时,都是在电子能量构成的势能面上进行的,因此IRC曲线的高度与反应能垒的含义完全不同,切不可从IRC曲线中“量取”能垒。
大多数简单反应的IRC曲线都与上述SN2类似,比较简单,但对于某些涉及多根化学键断裂/生成的反应,IRC曲线的形状会比较复杂。例如图6.3的IRC曲线中,在后过渡态区域先呈现出平缓的趋势,随后能量又突然下落。这对应了反应过程中多根化学键的变化并不是同步的。在过渡态中,只有C—Ag键发生了断裂,而在后过渡态区域的平坦位置,骨架开始发生重排,一根C—C键开始断裂;随后随着新的C—C键的形成,能量开始急剧下降。这类特征被称为协同非同步特性,在多键反应中很重要,而对IRC的研究则是最重要的研究方法。
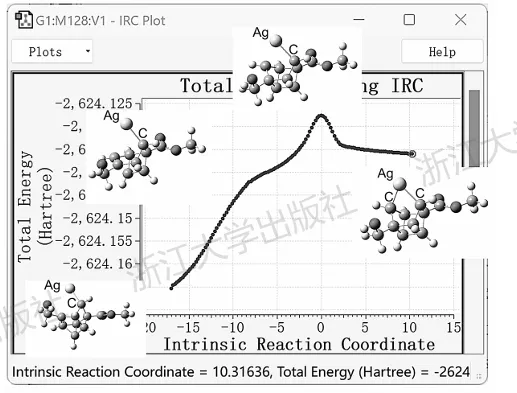
图6.3一个多键反应过渡态的IRC及其中部分结构
在对反应过程中的每个中间体和过渡态进行构型优化、频率和单点计算并得到自由能后,即可绘制出反应的自由能面。通常,习惯用“int”“im”等缩写命名中间体,“ts”命名过渡态。在这一部分,我们介绍如下几个方面来使得读者能清晰地认识自由能面。
首先介绍通过复杂反应的自由能面得到表观能垒的方法。
想要得到整个反应的能垒,即表观能垒,可以遵从如下步骤:
(1)将反应过程分阶段,每当遇到一个中间体能量低于前一个阶段的最低点时,就认为在此处进入一个新的阶段。
(2)在每个阶段内,取能量最低的中间体和能量最高的过渡态的自由能之差。
(3)在各阶段中,取上述差值最大者,即为该反应的表观能垒。
以图6.4中展示的自由能面为例,在Int3H处划分出了一个新阶段,两个阶段中能量最低和最高点之差分别为16.1kcal/mol和20.5kcal/mol,因此后者为本反应的表观能垒。这一反应的表观能垒由Int3H和TS3决定。
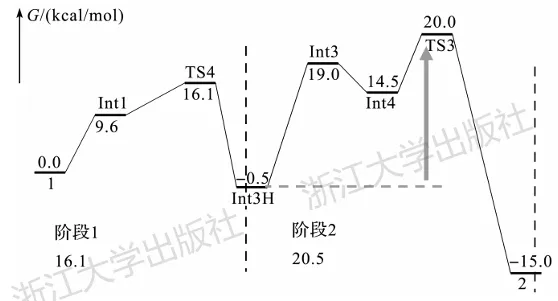
图6.4一个复杂反应势能面
随后介绍几个重要的概念。
1.resting state
当反应过程中存在某些低能中间体决定了能垒时,它们被称为resting states。图6.4
中的Int3H就是一个resting state。其越稳定,该反应的能垒就越高。从这个意义上来看,resting state对于高活性反应的开发是有害的。当计算结果告诉我们反应过程中存在resting state时,在后续反应的改良过程中,往往需要设法避免resting state的生成。
2.决速态(rate-determining state)
由上述例子可以知道,习惯上常说的“决速步”的概念并不总是存在。在上述势能面中,没有任何一个单一基元步骤的能垒决定了表观能垒。在这种情形下,比较适宜的说法是TS3是一个决速态。
3.预活化复合物(pre-activation complex)
在展示势能面时,我们总是以单独存在的各反应物作为自由能的零点。而在反应过程中,各反应物需要首先从无穷远处接近,往往能够形成一些复合物,它们被称为预活化复合物。在前文展示的SN2反应中,卤代烃与卤离子形成的复合物就是典型的预活化复合物。
由于结合过程面临不利的熵,在绝大多数情况下,预活化复合物相对于分立底物的自由能都是正的,因此不会对表观能垒进行影响,人们不会特意加以研究。而当预活化复合物构成一个resting state时(图6.5(b)),则需要将其找到,从而得到正确的表观能垒。
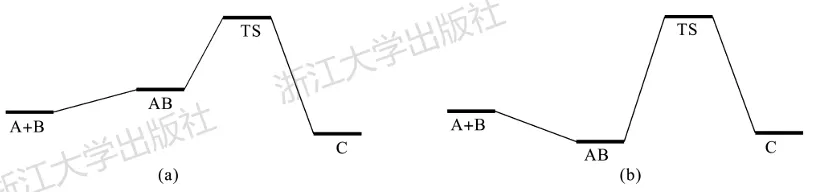
图6.5预活化复合物在势能面上的两种情形
4.无能垒反应和没有过渡态的反应
并非所有反应都存在过渡态。不存在过渡态的反应有如下两种类型:
(1)从原理上就不存在过渡态。这类反应的代表是电子转移反应。电子转移反应不遵从过渡态理论,其速率可通过Marcus理论加以描述。Marcus理论虽然可以描述电子转移反应的速率,但无法对反应过程中的结构变化带来信息,并且其中需要的重组能也不容易计算。加之大部分热力学允许的电子转移反应都比较快,很多情况下,对于电子转移反应,为了方便人们只研究热力学。
(2)反应过程中能量单调变化。过渡态是反应坐标上的极大值点,过渡态前后能量分别上升和下降。要让过渡态存在,反应过程中必然会同时存在能让能量上升的因素和能让能量降低的因素。在一些反应中,其中一种因素比另一种大得多,使得能量随着反应坐标发展而单调变化。这类反应的代表是正负离子结合、自由基结合等,在这些过程中只有化学键的生成这一使得能量下降的因素,而没有使得能量上升的因素。同理,作为其逆反应的化学键异裂和均裂等也没有过渡态。一些质子转移反应、激发态的反应等也经常是无能垒的。这些反应被统称为无能垒(barrierless)反应。对于放热的无能垒反应,可以认为反应速率受到扩散控制,反应物一经接近立刻反应;而对于吸热的无能垒反应,可以将其自由能变等价为能垒。
5.过渡态的自由能低于底物或产物的现象
对于某些反应,会发现过渡态的自由能低于底物或产物。根据其定义,过渡态是反应坐标上能量的极大值点,由于只有自由能有实际意义,此处的“能量”所指的同样是自由能。当实际结果发现过渡态能量低于底物时,有两种可能:
(1)存在预活化复合物。
(2)在构型优化过程中,实际上寻找的是电子能量势能面上的鞍点,随后通过频率计算得到的自由能校正量得到自由能。对于某些能垒极低的反应,这一项足以导致电子能量面和自由能面形状不同(这一项最主要的贡献者是熵,即这类反应的能垒主要由活化熵而非活化焓贡献),导致过渡态虽然是电子能量面上的鞍点,却不再是自由能面上的鞍点。这种情况下,此类反应可以当作无能垒。

多位专家力荐 超全实战指南


↑扫码了解更多书籍及唯理计算信息








