









 您已经拒绝加入团体
您已经拒绝加入团体

 2021-12-20
2021-12-20
 5604
5604
 0
0
【摘要】 若想用一个值衡量整体的结构改变程度,通过均方根偏差(Root-mean-square deviation,RMSD)值来评估是最恰当也是最省事的。
若想用一个值衡量整体的结构改变程度,通过均方根偏差(Root-mean-square deviation,RMSD)值来评估是最恰当也是最省事的。RMSD的定义如下:
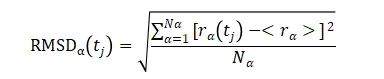
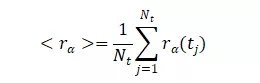
其中,Nα是用于和其位置相比较的一系列原子;Nt是用于比较一系列时间步骤内的原子位置;rα(tj)是α原子在时间tj时的位置;而< rα>表示α原子与rα(tj)位置间的平均值。
此处将通过免费软件VMD来演示如何将两个结构进行叠合,并计算RMSD值。
1.载入蛋白质
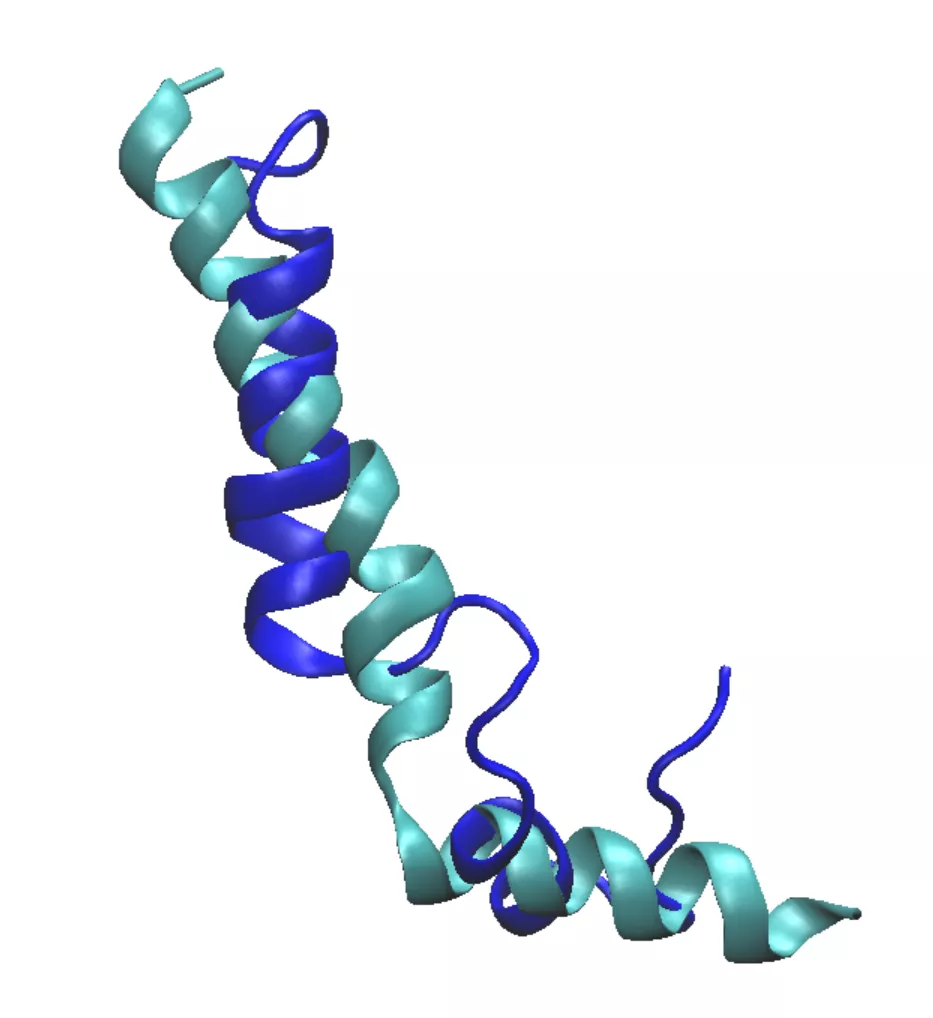
将模板蛋白质和目标蛋白载入到VMD窗口中。为直观比较,将两个结构采用不同的颜色进行绘制(具体可点击往期推文链接前往查看![]() VMD-分子结构可视化)。可以看到此时结构并未对齐。
VMD-分子结构可视化)。可以看到此时结构并未对齐。

2.叠合
选择Extensions - Analysis - RMSD Calculator,将左上角文本框内的内容改为all,点击Align,在图形窗口可看到两结构已经高度叠合。
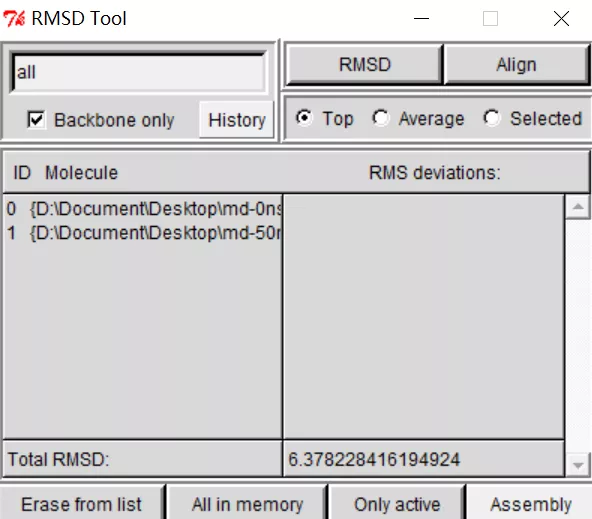
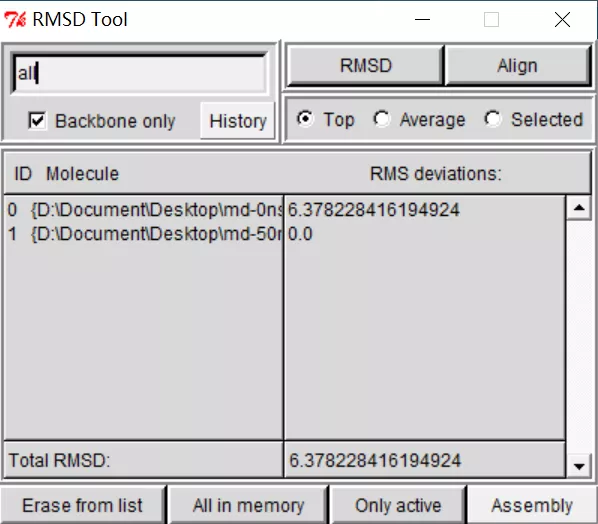
3.计算RMSD
点击RMSD Tool窗口中的RMSD选项,显示出两结构间的RMSD值。当前窗口看到的情况如下所示,可见RMSD是6.378Å。
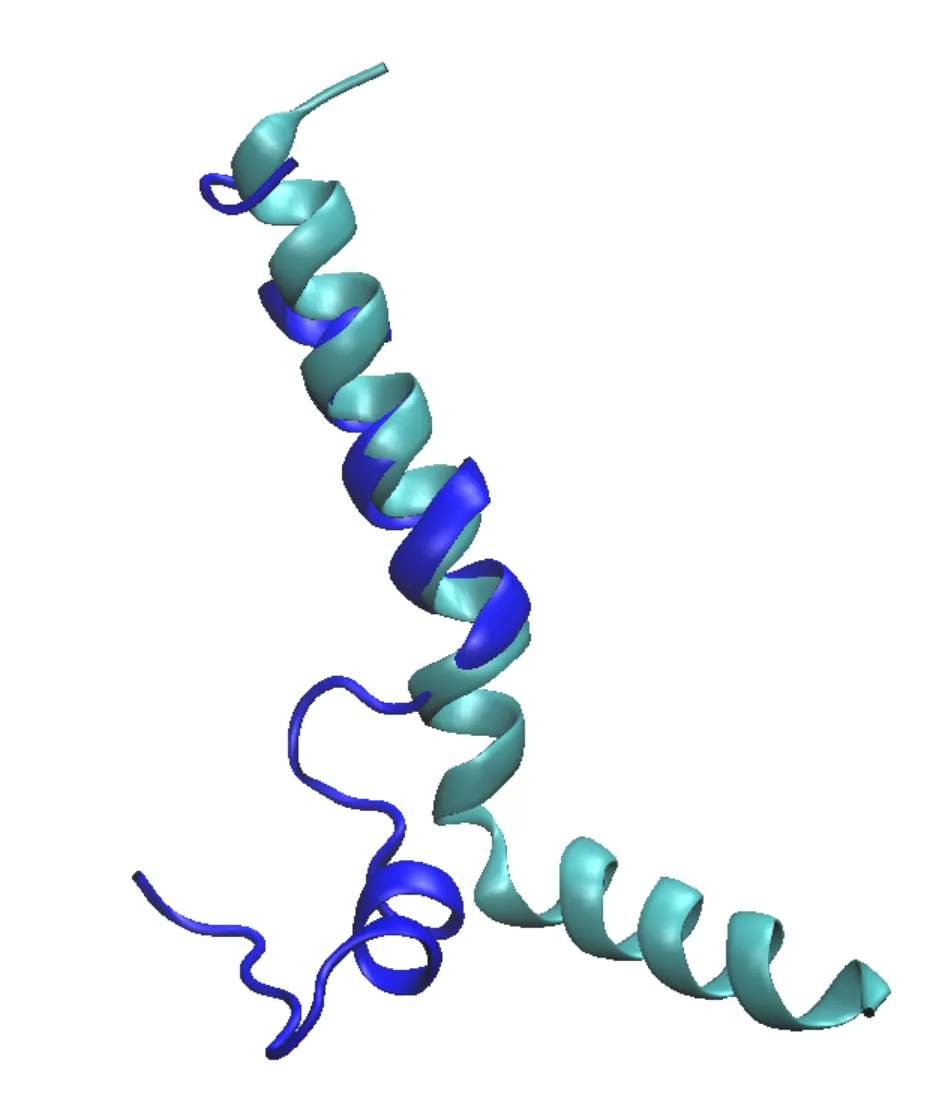
4.局部叠合
也可对蛋白的部分区域进行叠合和计算RMSD。例如,想对氨基酸16-21区域进行叠合时,在文本框内输入residue 15 to 20,叠合并计算,RMSD值变为1.979Å。

本文所有内容文字、图片和音视频资料,版权均属科学指南针网站所有,任何媒体、网站或个人未经本网协议授权不得以链接、转贴、截图等任何方式转载。








