









 您已经拒绝加入团体
您已经拒绝加入团体

 2022-10-08
2022-10-08
 3696
3696
 0
0
【摘要】 时至今日,计算化学早已不仅仅是实验的附属品了,它随着计算水平的逐步提升和理论框架的逐步完善,现在,越来越多的纯计算可以单独发在以往的顶刊之上了。
1.Angew:DUV NLO材料的一般原理:π共轭约束使带隙增大

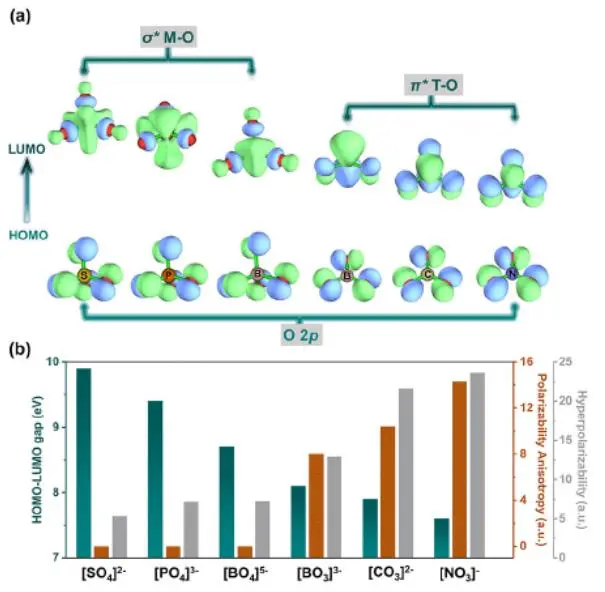
目前,非线性光学材料在带隙和双折射之间的权衡,面临着限制,特别是在深紫外光谱区域。在此,来自北京师范大学的陈玲等研究者为了解决这一难题,提出了一个普遍的原理,即π共轭约束,通过非π共轭单元的分离,使单元间的π共轭相互作用部分解耦。其目的是进一步扩大带隙,使其比奇异共轭共轭的带隙更大,并保持适当的π共轭单元密度,以获得较大的光学各向异性。研究表明,π共轭约束是目前所有DUV NLO材料的共同结构特征,为今后的设计合成,提供了新的、必要的设计准则。在这一原理的指导下,碳磷酸盐有望成为一种新的DUV候选体系。Sr3Y[PO4][CO3]3(1)和Na3X[PO4][CO3] (X = Ba, Sr, Ca, Mg, 2-5),不仅表现出比奇异磷酸盐大3 - 24倍的双折射率增强,还表现出比奇异碳酸盐岩宽0.2-1.7 eV的禁带增强。
参考文献:
Chen, L., Xiong, L. and Wu, L. (2021), A General Principle for DUV NLO Materials: π-Conjugated Confinement Enlarges Band Gap. Angew. Chem. Int. Ed.. Accepted Author Manuscript.
https://doi.org/10.1002/anie.202110740
原文链接:
https://onlinelibrary.wiley.com/doi/10.1002/anie.202110740
2.AM:在扭曲石墨碳氮化合物双层中的超快层间电荷分离,增强可见光吸收和可调谐过电势用于水分裂

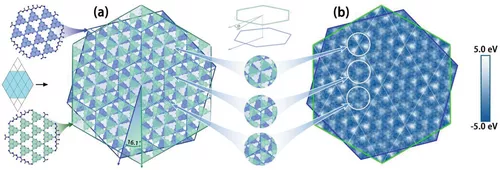
二维范德华层状结构,所形成的Moiré模式超晶格,因其广泛的应用而备受关注。在实验中,已经报道了双扭曲层体系对催化性能的提高,但其机理尚不清楚。在此,来自南京理工大学的陆瑞峰等研究者,从高精度第一性原理和含时从头算非绝热分子动力学计算,在120 fs内的超快层间电荷转移,优秀的电荷分离,改进的可见光吸收,在扭曲石墨氮化碳(g-C3N4)双层膜中发现了令人满意的析氢和析氧反应过电位,这有利于光催化、光电催化或电催化水裂解。该工作,为基于扭曲层状材料的先进纳米催化技术提供了指导。
参考文献:
Zhang, X., Wu, T., Yu, C., Lu, R., Ultrafast Interlayer Charge Separation, Enhanced Visible-Light Absorption, and Tunable Overpotential in Twisted Graphitic Carbon Nitride Bilayers for Water Splitting. Adv. Mater. 2021, 2104695.
https://doi.org/10.1002/adma.202104695
原文链接:
https://onlinelibrary.wiley.com/doi/10.1002/adma.202104695
3.Nano Letters:石墨烯中缺陷相关的波纹
石墨烯本质上的波纹和褶皱拓扑,从根本上影响其电子、力学和化学性质。实验技术,允许对原始石墨烯进行操作,并控制缺陷的产生,从而控制原子的平面外波动,从而调整石墨烯的特性。在此,来自英国剑桥大学的Angelos Michaelides等研究者,进行了大规模机器学习驱动的分子动力学模拟,以了解缺陷对石墨烯结构的影响。研究发现,缺陷引起明显更高的波纹,导致强烈的皱纹表面。这种结构转变的大小,强烈地依赖于缺陷浓度和特定类型的缺陷。分析缺陷的原子邻域,发现这些形态变化的程度,取决于缺陷的首选几何取向和缺陷之间的相互作用。虽然该研究强调了缺陷可以强烈影响石墨烯的形态,但它也通过将缺陷的整体结构与局部环境联系起来,强调了不同类型之间的差异。

参考文献:
Fabian L. Thiemann, Patrick Rowe, Andrea Zen, Erich A. Müller, and Angelos Michaelides, Defect-Dependent Corrugation in Graphene. Nano Letters Article ASAP DOI: 10.1021/acs.nanolett.1c02585
原文链接:
https://pubs.acs.org/doi/10.1021/acs.nanolett.1c02585
4.ACS Catalysis:复杂反应网络下氧化锌表面合成气转化产物选择性及结构演变研究

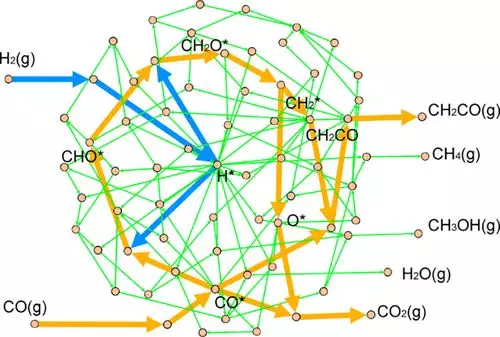
近年来,一种能选择性地将合成气转化为轻烯烃的双功能氧化沸石催化剂,得到了广泛的实验研究。OX-ZEO性能优异,但其机理存在争议。在此,来自中国科学院大连化学物理研究所的肖建平等研究者,首先开发了一种基于图论的算法,建立了一个完整的合成气转化为甲醇、烯酮和甲烷的反应网络。
研究者结合密度泛函理论(DFT)计算,利用反应相图系统研究了,合成气在氧化锌(ZnO)上的转化活性和选择性。实验中发现的关键中间体烯酮,在理论上首次得到了证实。氧化锌表面的演化,是影响产物选择性的关键因素。结果表明,低氧空位浓度的氧化锌表面更易产生甲醇。随着氧空位的增加,主要产物逐渐由甲醇演变为烯酮,最后演变为甲烷。因此,总的反应活性也增加了。研究者通过相图的预测,最后通过微观动力学建模进行了验证。
参考文献:
Xiaoyan Fu, Jiayi Li, Jun Long, Chenxi Guo, and Jianping Xiao. Understanding the Product Selectivity of Syngas Conversion on ZnO Surfaces with Complex Reaction Network and Structural Evolution. ACS Catalysis 0, 11. DOI: 10.1021/acscatal.1c02111
原文链接:
https://pubs.acs.org/doi/10.1021/acscatal.1c02111
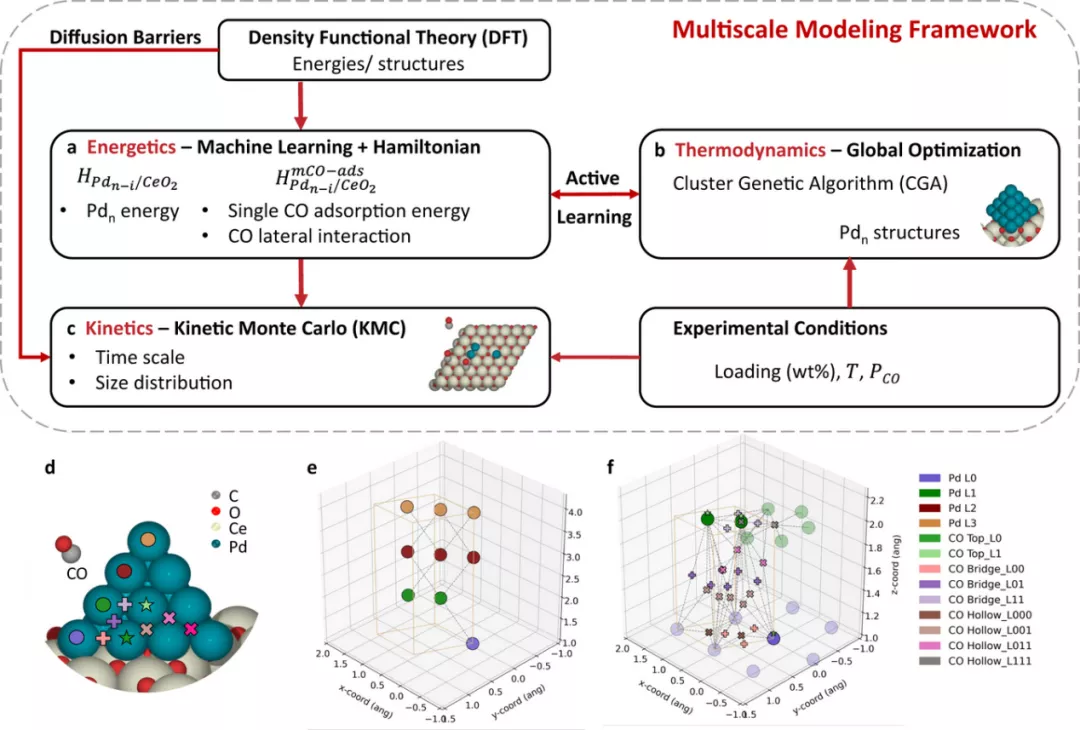
5.Nature Communications:基于多尺度模拟的负载亚纳米催化剂的实时动力学和结构

了解亚纳米催化剂的性能,以及催化剂处理和暴露在光谱探针分子中如何改变其结构,需要在工作条件下进行准确的结构测定。实验缺乏同时的时间和空间分辨率,可能会改变结构,类似的挑战阻碍了第一性原理计算来回答这些问题。
在此,来自荷兰埃因霍温理工大学的Emiel J. M. Hensen & 美国特拉华大学的Dionisios G. Vlachos等研究者,引入了一个多尺度模型框架来跟踪亚纳米簇在实验相关时间尺度上的演化。研究者证明了在不同催化剂负载、温度和CO暴露条件下,Pd吸附在CeO2(111)上的可行性。研究者表明,即使在室温下,烧结也在几秒钟内发生,主要是由自由能降低驱动的。它导致了准二维结构的动力学(远离平衡)冻结整体,这是一氧化碳化学吸附和红外实验所探测的。CO吸附使结构更平坦、更小。高温促使非常迅速的烧结向较大的,稳定/亚稳态的平衡结构,其中CO诱导二级结构的变化。
参考文献:
Wang, Y., Kalscheur, J., Su, YQ. et al. Real-time dynamics and structures of supported subnanometer catalysts via multiscale simulations. Nat Commun 12, 5430 (2021). https://doi.org/10.1038/s41467-021-25752-8
原文链接:
https://www.nature.com/articles/s41467-021-25752-8
本文所有内容文字、图片和音视频资料,版权均属科学指南针网站所有,任何媒体、网站或个人未经本网协议授权不得以链接、转贴、截图等任何方式转载。








