









 您已经拒绝加入团体
您已经拒绝加入团体

 2022-02-21
2022-02-21
 3331
3331
 0
0
【摘要】 玻璃成形能力作为合金一种重要的主要特性,但人们对其了解甚少,对其的量化在实验和计算上都很困难。
1.Nature Materials:数据驱动的金属玻璃成型能力通用指标的发现

玻璃成形能力作为合金一种重要的主要特性,但人们对其了解甚少,对其的量化在实验和计算上都很困难。在此,来自中国科学院物理研究所的Yan-Hui Liu等研究者,揭示了合金的玻璃形成能力表现在其远离平衡的非晶结构上,这可以通过常规X射线衍射来显示。具体来说,研究者从12个合金体系中制备了大约5700种合金,并对X射线衍射图中第一个衍射峰的半最大值Δq的全宽进行了表征。高的玻璃形成能力和大的Δq之间有很强的相关性。这种相关性表明,组成非晶结构的结构单元的大分散性是形成高金属玻璃的通用指标。与目前最先进的组合方法相比,当与组合合成配对时,相关性可提高100倍的吞吐量,并将有助于块状金属玻璃的发现。
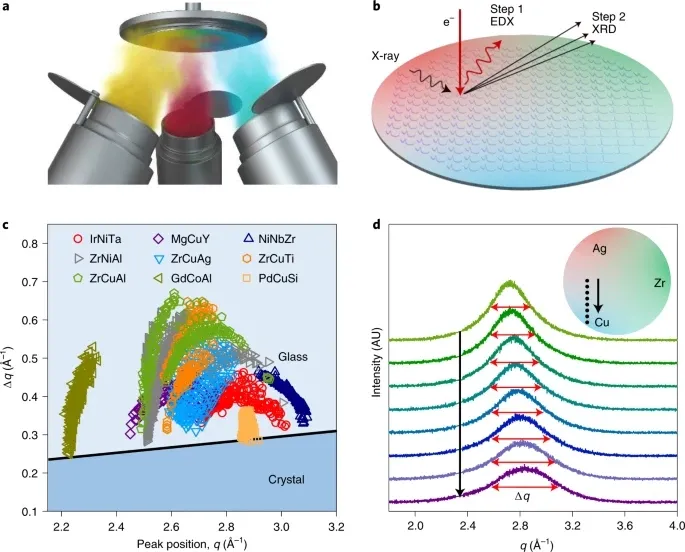
参考文献:
Li, MX., Sun, YT., Wang, C. et al. Data-driven discovery of a universal indicator for metallic glass forming ability. Nat. Mater. (2021). https://doi.org/10.1038/s41563-021-01129-6
原文链接:
https://www.nature.com/articles/s41563-021-01129-6
2.Nature Communications:通过密度修正的多体形式主义将密度泛函理论提升到水模拟的化学精度

密度泛函理论(DFT),已被广泛用于模拟水的性质。尽管在精度和效率之间保持了良好的平衡,但迄今为止,还没有密度泛函能够达到正确预测整个相图中水的性质所必需的精度。在此,来自美国加州大学圣地亚哥分校的Francesco Paesani等研究者提出了水的密度-修正扫描(DC-SCAN)计算,最大限度地减少密度驱动的误差,将扫描函数的精度提高到“金标准”耦合簇理论的精度。基于多体形式下的DC-SCAN的准确性,研究者引入了一个数据驱动的多体势能函数,MB-SCAN(DC),定量地复制耦合的水团相互作用、结合和单个多体能量的簇参考值。重要的是,MB-SCAN(DC)进行的分子动力学模拟也重现了液态水的性质,这表明MB-SCAN(DC)是第一个有效的基于DFT的模型,正确地描述了从气相到液相的水。
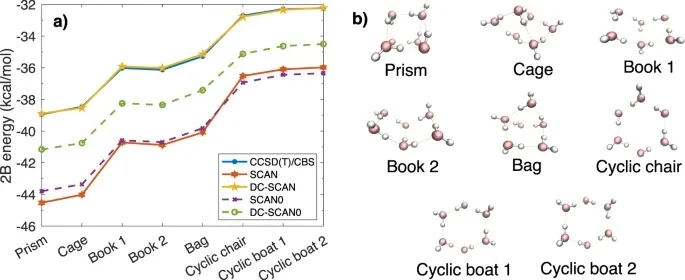
参考文献:
Dasgupta, S., Lambros, E., Perdew, J.P. et al. Elevating density functional theory to chemical accuracy for water simulations through a density-corrected many-body formalism. Nat Commun 12, 6359 (2021).
https://doi.org/10.1038/s41467-021-26618-9
原文链接:
https://www.nature.com/articles/s41467-021-26618-9
3.JACS:利用机器学习预测共轭寡电解质分子的抗菌活性

为了对抗日益增长的抗生素耐药性,需要新的抗生素,但是,从药物到先导,最终到一种有用的药物的开发过程需要几十年的时间。尽管利用机器学习方法进行分子属性预测的进展,为帮助抗生素开发过程开辟了新途径,但许多现有的解决方案依赖于大量数据集,并寻找与现有抗生素的结构相似性。在建立非传统抗生素分类模型方面仍然存在挑战,这些分类引起了越来越多的研究关注。
在此,来自美国麻省理工学院的Armi Tiihonen & Tonio Buonassisi和新加坡南洋理工大学的Sarah J. Cox-Vazquez & Guillermo C. Bazan等研究者,开发了一种共轭寡电解质分子的抗菌活性预测模型,这是一类新的且之前对此缺乏广泛的结构-活性关系研究的抗生素。该方法使得研究者能够预测大肠杆菌K12的最小抑制浓度,通过递归消除从一组5305个描述符中选择21个分子描述符。在不了解潜在机制的情况下,该预测模型的R2为0.65。该发现该结构域的最优分子表征是良好预测抗菌活性的关键。在共轭低电解质的情况下,反映分子三维形状的表示法是最关键的。虽然它是由一个共轭低电解质的具体例子证明的,但研究者提出的建立预测模型的方法,可以很容易地适应于其他新的抗生素候选域。
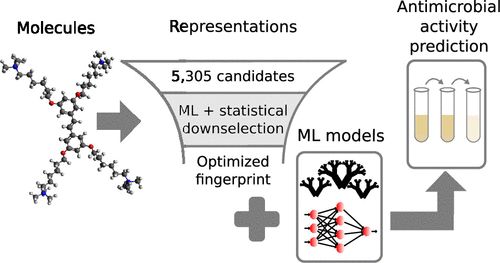
参考文献:
Tiihonen, Armi, et al. "Predicting antimicrobial activity of conjugated oligoelectrolyte molecules via machine learning." J. Am. Chem. Soc. 2021.
https://doi.org/10.1021/jacs.1c05055
原文链接:
https://pubs.acs.org/doi/10.1021/jacs.1c05055
4.ACS Catalysis:十聚钨酸镍催化脂肪族Csp3-H键芳基化与烷基化的机理研究

在此,来自沙特阿拉伯阿普杜拉国王科技大学的Bholanath Maity & Magnus Rueping & Luigi Cavallo等研究者,通过对十钨酸镍催化脂肪族Csp3-H键的芳基化反应进行计算分析,验证了反应机理,并对如何完成相应的烷基化反应提供了有价值的见解。该分析表明,光激发的十钨酸盐光催化剂通过一个涉及十钨酸盐O原子桥接的氢原子转移反应激活一个Csp3-H键。生成的烷基自由基与联吡啶基镍催化反应生成NiI-烷基中间体。芳基溴的氧化添加到这个中间产物导致了NiIII复合物的生成,通过还原消除将交叉耦合产物从其中解放出来。研究者还研究了相应的原型反应,其中芳基溴被一个烷基溴取代,这表明烷基溴氧化加成到NiI-烷基中间体的能量势垒过高。联吡啶配体的空间和电子性质的变化表明,6,6 '位置的例题累赘可以促进烷基溴的氧化加成,这已由专门的实验验证。
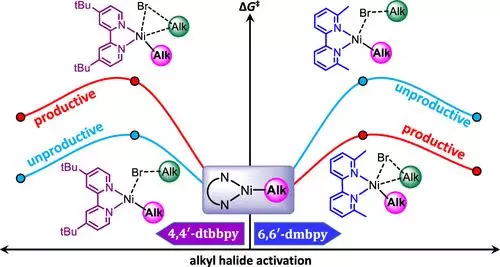
参考文献:
Maity, Bholanath, et al. "Mechanistic Understanding of Arylation vs Alkylation of Aliphatic Csp3-H Bonds by Decatungstate-Nickel Catalysis." ACS Catalysis 11 (2021): 13973-13982.
原文链接:
https://pubs.acs.org/doi/10.1021/acscatal.1c04142
5.ACS Nano:宽高比大于1000的Cu纳米线的固相生长:多尺度论

五孪金属纳米线,在现有技术和新兴技术中得到了广泛的应用。然而,人们对它们的生长机制知之甚少。在此,来自美国宾夕法尼亚州立大学的Kristen A. Fichthorn等研究者,利用多尺度理论从第一性原理探讨了氯和烷基胺介导的五孪铜纳米线的溶液-相生长的起源。利用量子密度泛函理论(DFT)计算,研究者描述了铜原子在氯覆盖的Cu(100)和Cu(111)表面的结合和表面扩散。研究发现,在氯化Cu(111)表面上,Cu原子的结合强度比在氯化Cu(100)表面上更强,扩散速度更慢,这与裸铜表面的趋势相反。研究者还分析了面间扩散,发现从Cu(100)到Cu(111)的扩散速度要快于从面间扩散到Cu(111)的速度。在埃级尺度上,使用DFT在单个位点之间跳跃的速率,研究者计算了不同长度──可达数百微米──和直径在10纳米范围内的纳米线的粗粒度、界面间的速率。研究者仅根据表面扩散,预测纳米线的纵横比为~ 100。研究者还考虑了自组装烷基胺层的影响,该层覆盖了大多数{100}面,但在{111}面和靠近{100}/{111}边界的“端区”上缺乏或薄且无序。通过端区,研究者预测了在实验范围内的纳米线宽高比。该工作揭示了卤化物促进高纵横比纳米线生长的机制。
参考文献:
Kim, Junseok, Jianming Cui, and Kristen A. Fichthorn. "Solution-Phase Growth of Cu Nanowires with Aspect Ratios Greater Than 1000: Multiscale Theory." ACS nano (2021).
https://doi.org/10.1021/acsnano.1c07425
原文链接:
https://pubs.acs.org/doi/10.1021/acsnano.1c07425
本文所有内容文字、图片和音视频资料,版权均属科学指南针网站所有,任何媒体、网站或个人未经本网协议授权不得以链接、转贴、截图等任何方式转载。








