









 您已经拒绝加入团体
您已经拒绝加入团体

 2022-03-17
2022-03-17
 3776
3776
 0
0
【摘要】 时至今日,计算化学早已不仅仅是实验的附属品了,它随着计算水平的逐步提升和理论框架的逐步完善,现在,越来越多的纯计算可以单独发在以往的顶刊之上了。
时至今日,计算化学早已不仅仅是实验的附属品了,它随着计算水平的逐步提升和理论框架的逐步完善,现在,越来越多的纯计算可以单独发在以往的顶刊之上了。
1.Nature Communications:通过高通量计算筛选加速超氧化物歧化酶纳米酶的发现

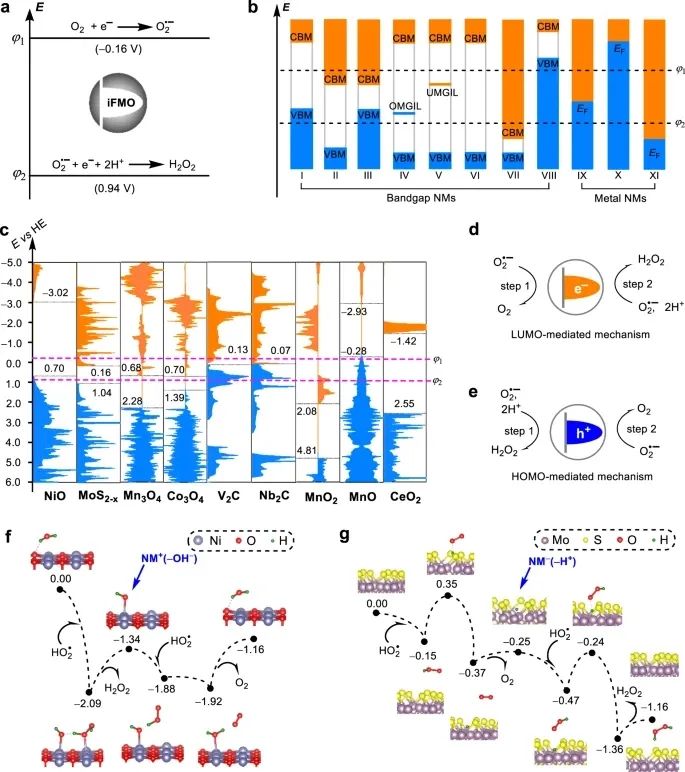
纳米材料(NMs)对超氧阴离子的催化清除活性与超氧化物歧化酶(SOD)相似。虽然已有数十种超氧化物歧化酶,被证明具有这种活性,但其基本原理尚不清楚,从而阻碍了超氧化物歧化酶作为新型超氧化物歧化酶的发现。在此,来自中国国家纳米科学与技术中心的 Xingfa Gao等研究者,利用密度泛函理论计算来研究了催化过程的热力学和动力学,从而发展了可用于活性的两个规则,即一个能级规则和一个吸附能量规则。第一原理定量地描述了中间前沿分子轨道在催化电子转移中的作用。第二部分定量地描述了期望的催化反应和非期望的副反应之间的竞争。实验验证了该原理预测金属有机骨架类SOD活性的能力。这两个原理都可以很容易地在计算机程序中实现,以计算筛选具有固有的超氧化物歧化酶活性的NMs。
参考文献:
Wang, Z., Wu, J., Zheng, JJ. et al. Accelerated discovery of superoxide-dismutase nanozymes via high-throughput computational screening. Nat Commun 12, 6866 (2021). https://doi.org/10.1038/s41467-021-27194-8
原文链接:
https://www.nature.com/articles/s41467-021-27194-8
2.Nature Computational Science:纳米多孔超级电容器恒流充放电的建模

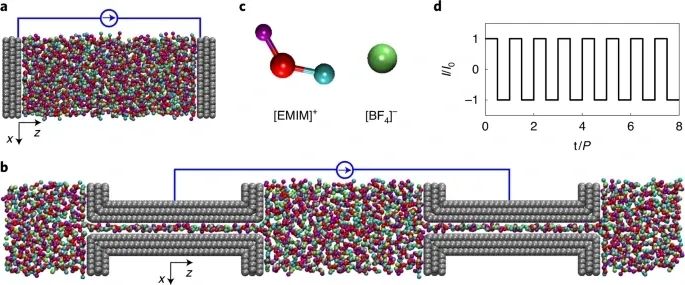
要想从原子水平上研究超级电容器的能量储存,分子模型是必不可少的。恒电位法(CPM),可以使电极内的电势保持均匀,这对于真实地描述超级电容器中的电荷再分配和动力学过程至关重要。然而,以往的CPM研究仅限于恒电位模式。恒电流模式虽然被广泛应用于实验中,但由于缺乏有效的方法,在CPM模拟中很少被研究。在此,来自华中科技大学的Guang Feng等研究者,提出了一种模拟恒电位下超级电容器恒电流充放电过程的建模方法。研究表明,对于纳米多孔电极,这种建模方法可以捕获超级电容器中与实验一致的动态。在分子尺度上,它还能反映出充放电过程中离子吸附-解吸动力学的滞后现象。这种方法使得超级电容器动力学中物理和电化学的进一步精确建模成为可能。
参考文献:
Zeng, L., Wu, T., Ye, T. et al. Modeling galvanostatic charge–discharge of nanoporous supercapacitors. Nat Comput Sci 1, 725–731 (2021). https://doi.org/10.1038/s43588-021-00153-5
原文链接:
https://www.nature.com/articles/s43588-021-00153-5
3.JACS:二氢化物和二氢配合物在单原子催化剂上析氢反应中的作用

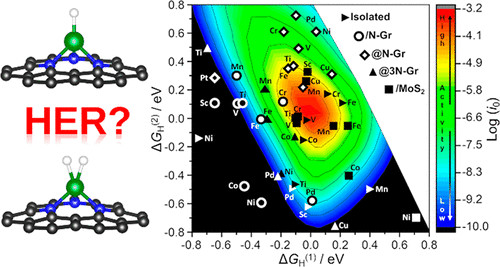
析氢反应(HER),在电化学水分解中起着关键作用。近年来,利用单原子催化剂(SACs)对HER,进行了大量研究。在HER中SACs的活性,通常是合理化或使用由Nørskov提出的原始模型,其中H原子吸附在扩展的金属表面M (MH中间体的形成)上的自由能用来解释HER交换电流的趋势。然而,SACs与金属表面有很大的不同,可以认为是配位化合物的类似物。在配位化学中,与金属表面不同,可以形成稳定的二氢化物或二氢配合物(HMH)。在此,来自意大利米兰比可卡大学的Gianfranco Pacchioni等研究者,研究表明,上述同样的情况也会发生在SACs上,并且会形成稳定的HMH中间体,除了MH中间体之外,可能会改变这个过程的动力学。将原来的动力学模型扩展到两种中间产物(MH和HMH)的情况下,得到了SACs上HER的三维火山图。对55个模型的DFT数值模拟表明,新的动力学模型可能对SACs在HER中的活性得出完全不同的结论。通过选定的实验实例验证了结果的正确性。这项工作,为SACs和配位化合物之间的化学相似性提供了一个重要的范例。
参考文献:
Di Liberto, Giovanni, Luis A. Cipriano, and Gianfranco Pacchioni. "Role of Dihydride and Dihydrogen Complexes in Hydrogen Evolution Reaction on Single-Atom Catalysts." J. Am. Chem. Soc. 2021.
https://doi.org/10.1021/jacs.1c10470
原文链接:
https://pubs.acs.org/doi/10.1021/jacs.1c10470
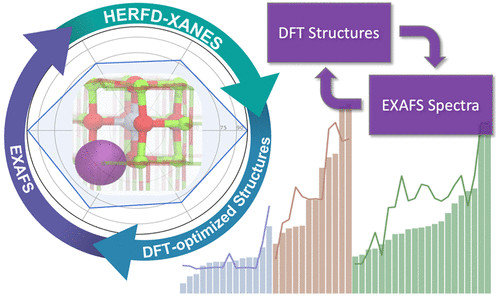
4.JACS:理论指导X 射线吸收光谱方法用于识别原子分散过渡金属催化剂中的活性位点

原子分散的负载金属催化剂,提供了新的性能和最大限度的金属可及性和利用的优点。然而,这些材料的表征仍然具有挑战性。在此,来自美国SLAC国家加速器实验室的Simon R. Bare & 美国加州大学的Coleman X. Kronawitter & Bruce C. Gates & Ambarish R. Kulkarni等研究者,使用原子分散的铂支撑晶体MgO(选择其定义良好的键合位点)作为一个典型的例子,演示了如何系统性地密度泛函理论计算来评估所有潜在稳定的铂位点,并结合扩展X射线吸收精细结构(EXAFS)光谱的自动分析,来无偏倚地识别孤立的、表面包膜的铂阳离子作为CO氧化的催化物种。采用原子分辨率成像、EXAFS和高能分辨荧光检测X射线吸收近边缘光谱对催化剂进行了表征。所提出的铂位点与实验结果一致。这种理论指导的工作流程,获得了严格确定的结构模型,并提供了比目前常规EXAFS分析更详细的催化活性位点结构的图片。由于这种方法对金属、载体和催化反应都是有效且不可知的,研究者认为它将在材料表征和催化领域中具有广泛的应用前景。
参考文献:
Chen, Yizhen, et al. "A Theory-Guided X-ray Absorption Spectroscopy Approach for Identifying Active Sites in Atomically Dispersed Transition-Metal Catalysts." J. Am. Chem. Soc 2021.
https://doi.org/10.1021/jacs.1c07116
原文链接:
https://pubs.acs.org/doi/10.1021/jacs.1c07116
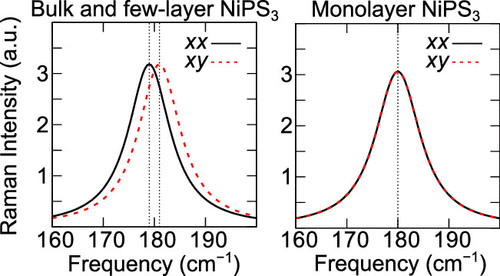
5.Nano Letters:过渡金属三硫化磷的磁各向异性和磁有序

在此,来自韩国首尔国立大学的Tae Yun Kim & Cheol-Hwan Park等研究者,通过对过渡金属三硫化磷(TMPS3)的第一性原理计算,确定了一个具有前所未有的大量参数的磁模型,再现了测量到的块状TMPS3的磁基态。研究者对TMPS3的临界温度、磁化率和比热的蒙特卡罗模拟与已有的实验数据一致,表明TMPS3的反铁磁序一直持续到单层。值得注意的是,在最近的第一性原理研究中被忽略的轨道极化,显著地提高了FePS3的磁各向异性近2个数量级。最近的拉曼研究[Kim, K.,Nat. Commun. 2019,10,345]声称单层NiPS3中不存在磁有序,但同时报道了一个强的双-磁振子连续体;研究者证明了用于判断单层NiPS3磁性有序的准则是无效的,因为它提供了两个看似矛盾的实验结果的理解。因此,对少层TMPS3的磁化率和比热的丰富预测,有待实验验证。
参考文献:
Kim, Tae Yun, and Cheol-Hwan Park. "Magnetic anisotropy and magnetic ordering of transition-metal phosphorus trisulfides." Nano Lett 2021.
https://doi.org/10.1021/acs.nanolett.1c03992
原文链接:
https://pubs.acs.org/doi/10.1021/acs.nanolett.1c03992
本文所有内容文字、图片和音视频资料,版权均属科学指南针网站所有,任何媒体、网站或个人未经本网协议授权不得以链接、转贴、截图等任何方式转载。








