









 您已经拒绝加入团体
您已经拒绝加入团体

 2024-12-30
2024-12-30
 5929
5929
 0
0
【摘要】 单网络电荷密度泛函可以预测硅、砷化镓、二硫化钼、锗、磷酸锡、磷酸钛和磷酸锆七种不同材料的高能电子能隙,其相对分子质量为172.6 meV,比标准的高能电子能隙回归精度高34%。
密度泛函理论(DFT)已成为从头计算材料性质的标准方法。然而,它也有一些缺点,特别是在预测依赖于激发态的关键性质,如带隙和光谱方面。为了处理这样的性质,可以采用更精确的方法,例如具有混合泛函的GW或DFT(仅举几例,包括HSE、PBE0和B3LYP);然而,由于其高计算成本和大存储需求,这些方法对于许多大型和/或复杂的系统是不可行的。
Levi C Lentz[1]等研究了用标准PBE泛函计算的传统DFT电荷密度训练神经网络以准确预测HSE带隙的能力。结果表明,单网络电荷密度泛函可以预测硅、砷化镓、二硫化钼、锗、磷酸锡、磷酸钛和磷酸锆七种不同材料的高能电子能隙,其相对分子质量为172.6 meV,比标准的高能电子能隙回归精度高34%。
这种方法原则上可以用来将PBE电荷密度映射到带隙或用任何更高精度的方法计算的其他性质,具有降低计算成本、提高预测精度的潜力,并能够对各种复杂材料系统进行准确的高通量筛选。
一种替代方案是采用混合交换,如Heyd-Scuseria-Ernzerhof (HSE)或Becke,三参数,Lee-Yang-Parr (B3LYP)。这些方法修改了PerdewBurke-Ernzerhof (PBE)交换能量与精确交换的部分,参数化以确保在特定数据集上的特定性能与实验一致。虽然这些方法获得了更好的结果,但它们在计算上也过于昂贵,无法用于高通量筛选或非常大的系统。降低这些方法的计算成本将使研究人员能够将这些方法应用于目前标准技术无法实现的更广泛的系统。
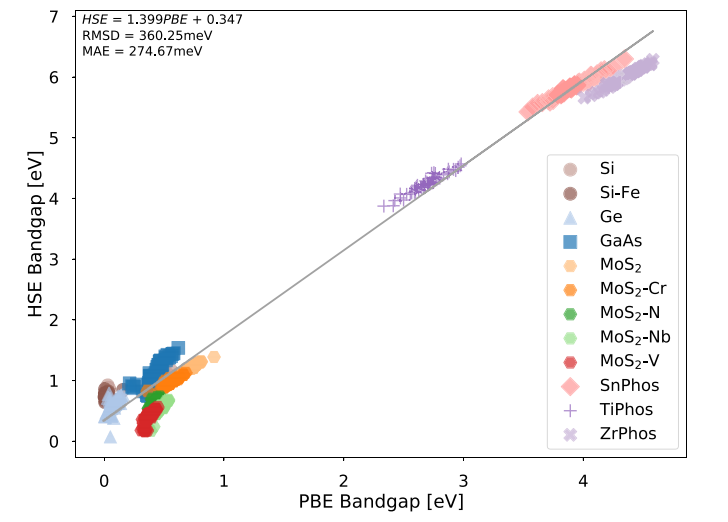
图1. HSE带隙与PBE带隙。[1]
Levi C Lentz[1]等探索使用机器学习方法,特别是神经网络来降低高精度计算的计算成本,以HSE计算为例,目标是高阶方法。他们利用这种机制,通过有效地学习预测HSE带隙所需的PBE电荷密度的功能,将PBE电荷密度映射到HSE带隙。虽然这种方法不能直接校正PBE电荷密度来解释自相互作用项,但它是一种强大的方法,可以通过对小数据集的训练,将计算成本低的方法(如PBE)映射到相对昂贵的方法(如HSE)。
在这项工作中,他们展示了神经网络在广泛的系统和物种中学习这种功能形式的能力;将这项工作扩展到其他材料特性可以使神经网络增强传统的DFT方法,以降低计算成本并提高准确性。独特的是,该方法不依赖于输入数据集的任何转换;学习到的网络函数存在于电荷密度空间中,这可能使对神经网络函数的分析能够更好地理解高阶方法如何纠正PBE等方法中的不准确性。
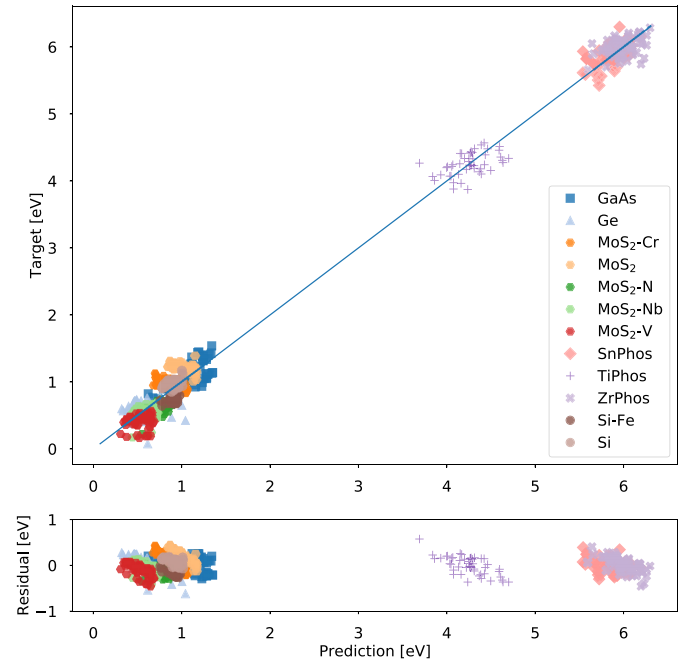
图2. 5个150神经元单层网络函数的平均输出。[1]
他们展示了神经网络泛函的潜在应用:解决PBE泛函在准确预测材料带隙方面的缺点。我们已经证明了使用PBE电荷密度作为神经网络的输入数据来学习PBE电荷密度与HSE带隙之间的关系的能力。
已经证明,神经网络可以胜过简单的统计方法,如线性回归。在这里优于线性回归表明神经网络函数可以大大提高非线性关系建模的准确性。这种方法可以帮助学习功能关系完全未知的关系。这可能是计算界的福音,因为这将显著降低依赖于精确DFT方法估计属性的计算成本。
[1] Levi C Lentz and Alexie M Kolpak 2020 J. Phys.: Condens. Matter 32 155901
科学指南针充分发挥互联网技术和业务优势,在国内率先打造出业界领先的线上化、数字化的科研服务基础设施,在行业内首创用户自主下单、服务全流程追踪、测试“云现场”等模式,进一步提高了大型科学仪器设施开放共享和使用效率,以实际行动助力科技创新。现已发展成为中国专业科研服务引领者,已获得检验检测机构资质认定证书(CMA)、实验动物使用许可证、“ISO三体系认证”等专业认证。
免责声明:部分文章整合自网络,因内容庞杂无法联系到全部作者,如有侵权,请联系删除,我们会在第一时间予以答复,万分感谢。








