









 您已经拒绝加入团体
您已经拒绝加入团体

 2021-09-24
2021-09-24
 34197
34197
 0
0
【摘要】 密度泛函理论(Density Functional Theory,DFT)作为处理多粒子体系的近似方法已经在凝聚态物理、材料科学、量子化学和生命科学等领域取得了广泛应用。
密度泛函理论(Density Functional Theory,DFT)作为处理多粒子体系的近似方法已经在凝聚态物理、材料科学、量子化学和生命科学等领域取得了广泛应用。例如,图1(c)是利用DFT方法计算的72个原子的超晶胞结构[1],基于DFT的材料科学计算模拟方法不仅可以研究现有材料,而且可以预测新材料。
图1
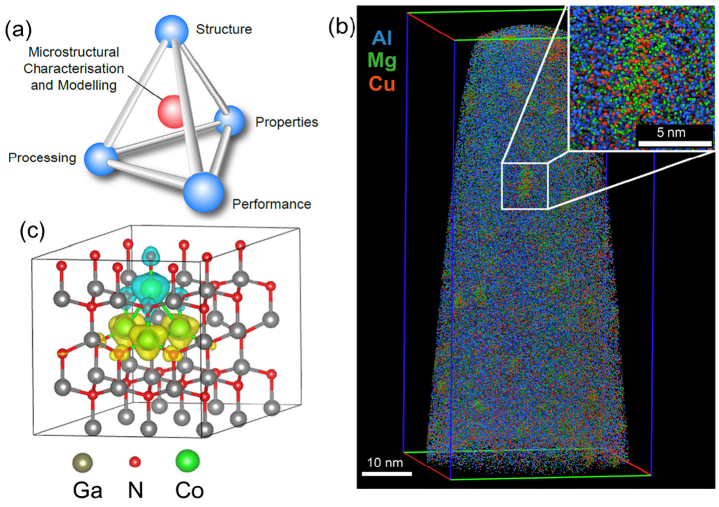
(a)显微表征、结构、加工、性质和性能之间的潜在联系,(b)约含2千万个原子的Al-Cu-Mg合金的APT数据,(c)DFT计算的72-atom超晶胞实例
泛函是从向量空间到标量的映射,即函数的函数。表1列出了一些已经提出的密度泛函的种类,其中有些是从基本量子力学推出的,有些是通过参数化函数得出的,都有各自的优缺点和适用范围[2]。DFT方法的实质是将电子密度作为分子(原子)基态中所有信息的载体,而不是单个电子的波函数,从而使多电子体系转化为单电子问题进行求解。假设电子数目为N,则波函数中的变量数为3N,而密度泛函理论将变量数缩减到三个空间变量,这样既简化了计算过程,又可以确保计算精度。
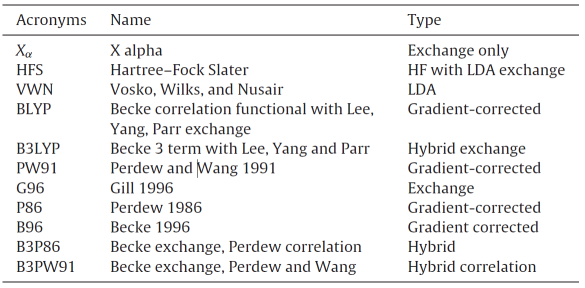
表1 一些近似的密度泛函种类
密度泛函理论发展至今大致可分为三个阶段。第一阶段是1927年,Thomas、Fermi基于理想状态下的均匀电子气假设提出了Thomas-Fermi 模型,第一次引入了密度泛函的概念,成为后来DFT方法的雏形。
Thomas-Fermi模型的出发点是假设电子之间无相互作用且无外力干扰,则电子运动的薛定谔方程可表示为:
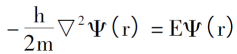 解得:
解得: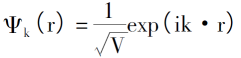
引入0K下的电子排布规律,电子密度、单电子总能和体系的动能密度分别为:
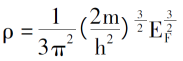
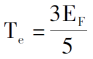
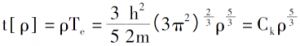
再引入对电子间的库伦势和外场的描述,可以推出仅由电子密度函数决定的电子体系的总能表达式[3]。
该模型虽然简化了计算形式和过程,却没有考虑电子间的交互作用,没有对动能项进行精确描述,所以在很多体系中并不适用。但相关学者在这一新颖的研究思路的启发下,经过多年的努力在20 世纪60 年代基本完善了密度泛函理论的内容,并最终建立起严格意义的密度泛函计算理论。
图2 基于DFT的自洽迭代流程示意图
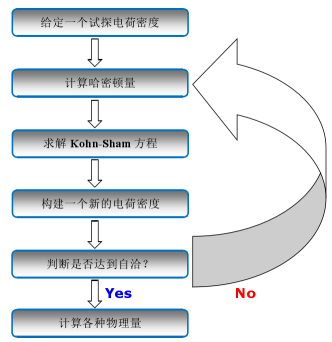
Hohenberg-Kohn 定理和Kohn-Sham方程的提出对DFT方法的形成和完善起到了关键作用,被誉为DFT的两大基石。
(1)Hohenberg-Kohn定理
Hohenberg-Kohn定理的核心思想是:体系中的所有物理量都可以通过只包含电子密度的变量来唯一决定,而实现方法是通过变分原理来求得体系基态。该理论主要针对非均匀电子气模型,并由两条子定理构成。i)处在外势( 除电子相互作用以外的势) 的忽略自旋的电子体系,其外势可通过电子密度唯一决定;ii)对于给定的外势,系统基态能量即能量泛函的最小值。于是,体系的能量泛函可描述为:
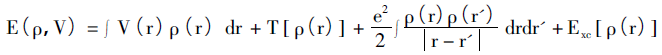
等式右边分别为外场中的电子势能、动能项、电子间的库伦作用和交换关联势能。
此定理并未给出电子密度函数、动能泛函和交换关联泛函的具体表达式,因此具体求解还是无法进行。
(2)Kohn-Sham方程
直到1965年,Kohn和沈吕九建立了Kohn-Sham方程,给出了各项的具体描述形式,使密度泛函理论开始进入实际应用阶段。他们针对动能泛函提出使用无相互影响的粒子动能泛函来近似代替,而将二者的差异纳入交换关联泛函的未知项中[4]。
定义密度函数为:![]() ,对ρ的变分采用Φi(r)的变分代替,拉格朗日乘子用Ei代替,则单电子方程为:
,对ρ的变分采用Φi(r)的变分代替,拉格朗日乘子用Ei代替,则单电子方程为:
其中,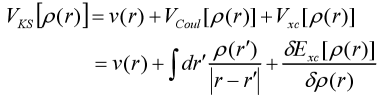
以上即为Kohn-Sham方程。
Kohn-Sham方程给交换关联泛函以外的各项都赋予了明确的表达形式,并把复杂作用项也归并在此项中。至此,计算难度得到大大简化,所有的工作都开始围绕着如何描述交换关联泛函展开,同时,交换关联势的近似形式也直接决定了密度泛函理论的精度。
局域密度近似(Local Density Approximation,LDA)方法也是由Kohn和沈吕九在1965 年提出的,目的是将未知的交换关联项近似表达,使DFT方法能够用于实际计算。LDA使用均匀电子气的密度函数来计算非均匀电子气的交换关联项,假设体系中的电子密度随着空间的变化极小,则非均匀电子气的交换关联项可以表示为:
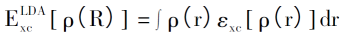
相应的交换关联势可表示为:
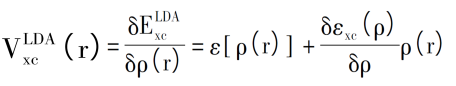
例如,Asad Mahmood等人使用VASP比较了LDA-PBE和GGA-PAW计算的平衡结构参数,研究了Ga掺杂对电子轨道杂化,以及光学性质和晶体几何形状的影响,从图3(c)可以看出Ga-2s和Ga-2p轨道对导带有显着贡献,较低的VB也由Ga-2p贡献,CB底部的杂质带提示了额外的能垒,在VB和CB之间的电子跃迁须越过该能垒[5]。
图3 计算结果
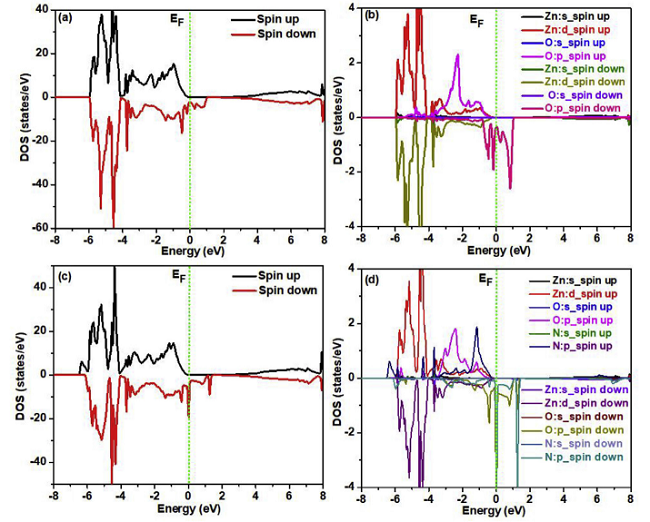
(a)优化的3x3x3 Ga掺杂ZnO超晶胞,(b)能带结构,(c)态密度DOS
为了更加精确地计算实际物质体系,1986年,Becke、Perdew和Wang等人提出了广义梯度近似(Generalized Gradient Approximation,GGA),这也是目前密度泛函计算中运用最为广泛的处理方法。
GGA 处理方法是将原来的表示项改写为包含电子密度和梯度函数的泛函形式,再加上对自旋的描述,得到的交换关联泛函如下:
![]()
在GGA中,交换关联势也可以拆解为交换能和关联能。那么,如何构建这两部分的合理表达式呢?Beckc 等人认为具体的泛函形式原则上可以任意构建,并不需要考虑实际的物理意义,比如GGA-PW91;而Perdew 等人则主张尽量回归到纯粹的量子力学计算理论,所有的物理量计算仅从电子静质量、普朗克常数、光速等基本常量入手,泛函表达式不应过多包含经验参数,例如在凝聚态物理学等领域经常使用的GGA-PBE(Perdew-Burke-Enzerhoff)。
最近,Parmod Kumar、L. Romaka等人分别利用WIEN2k和Elk v2.3.22中的FP-LAPW(全电势线性增强平面波)进行了相关研究,其中,交换关联势采用GGA-PBE形式,图4、5为相应的态密度和电荷密度分布等计算结果[6,7]。
图4 未注入和注入N的自旋极化ZnO超级电池的总态密度和局域态密度
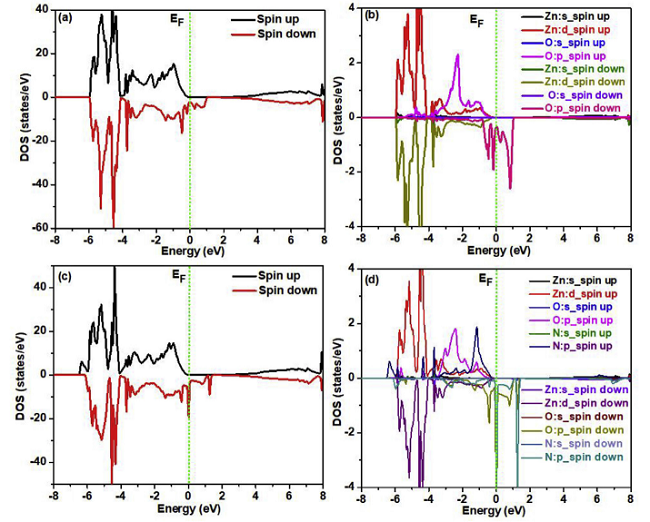
图5 VCoSb锑化物中电子定位函数(Y)和电荷密度(r)的分布
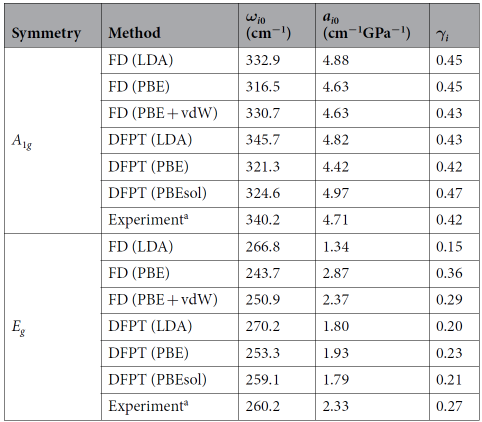
J.Ibáñez, T. Woźniak等人测试了不同密度泛函理论的有效性,以预测HfS2在压力下的晶格动力学性质。观察表2发现GGA-DFT恰当地描述了考虑vdW相互作用时HfS2的高压晶格动力学,而LDA-DFT计算广泛用于预测2D化合物在环境条件下的结构和振动特性,无法再现压缩条件下HfS2的行为,这表明使用DFT-LDA计算TMDCs的压缩性和Grüneisen参数会产生较大的误差[8]。
表2 拉曼频率(ωi0)、压力系数(ai0)和Grüneisen参数(γi)
除了LDA 和GGA两种算法外,还有以杂化的方式将Hatree-Fock(HF)交换作用影响纳入到交换关联项的杂化密度泛函,比如,在1998年很受欢迎的B3LYP等。这些理论包含的体系信息越来越多,计算结果也与试验数据越来越接近,尤其适用于有机化学领域,并在化学反应机理计算方面获得了很大的成功。
例如,T. Garwood等人使用PBE0型杂化计算了InAs/GaSb II型超晶格结构(模型如图6所示)的带隙数据[9],与试验值非常接近,偏差范围约为3%-11%。
图6 使用VESTA计算的杂化DFT的Ball-and-stick InAs / GaSb SLS模型
基于Kohn-Sham单粒子能谱的电子能带结构理论可以对很多材料进行定性描述,但从定量角度却不尽人意。例如,对于Si 、GaAs等简单的半导体材料,在LDA/GG下的Kohn-Sham DFT给出的带隙远远偏小;对于Ge、InN等小带隙半导体,从LDA/GGA得到的是金属态,而实验上观测到的却是半导体,这就是所谓的LDA/GGA的带隙问题。
为了克服带隙问题,人们在DFT的理论框架内做了大量的努力,比如把基于局域有效势的Kohn-Sham理论扩展到了基于非局域有效势的Generalized Kohn-Sham(GKS)理论,以及其它杂化密度泛函理论,还有一种基于单体格林函数(one-body Green’s function)的多体微扰理论。该理论中,与Kohn-Sham DFT的交换关联势相对应的是交换关联自能算符。对于自能算符,一个相对简单而准确的近似就是GW近似(单粒子格林函数G和屏蔽库仑作用W的乘积),通过计算一定近似下的自能算符,便可以得到对应于PES(IPS)中的准粒子激发能量。这些新的发展方向虽然改进了对材料带隙的描述,但近似泛函仍存在着很大的主观性,适用范围相对有限,至今还未有一个普适的、有充分理论基础的DFT方法实现对材料电子能带结构的准确描述[10,11]。
此外,还有一些在现有密度泛函理论基础上的扩展,比如,以KS轨道能量差为基础展开激发能的含时密度泛函理论(TDDFT),用单粒子Dirac方程代替Schordinger方程的相对论性密度泛函理论,扩展到强关联体系的LDA+U,以及处理任意强度磁场下相互作用电子体系的流密度泛函理论(CDFT)等。
参考文献
Xiang-Yuan Cui, Simon P. Ringer,On the nexus between atom probe microscopy and density functional theorysimulations [J] ,Materials Characterization (2018), https://doi.org/10.1016/ matchar.2018.05.015
B. Obot, D.D. Macdonald, Z.M. Gasem,Density functional theory (DFT) as a powerful tool for designing neworganic corrosion inhibitors. Part 1: An overview [J],Corrosion Science 99 (2015) 1–30
Geerlings, F. De Proft, W. Langenaeker, Conceptual density functional theory,Chem. Rev. 103 (2003) 1793–1873.
Nagy, Density functional theory and application to atoms and molecules, Rev. 298 (1998) 1–79.
Koch, M.C. Holthausen, A Chemist’s Guide to Density Functional Theory,Wiley-VCH, Weinheim, 2000.
Asad Mahmood, Fatih Tezcan, Gulfeza Kardas,Thermal decomposition of sol-gel derived Zn0.8Ga0.2O precursor-gel: Akinetic, thermodynamic, and DFT studies [J],Acta Materialia 146 (2018) 152-159
Parmod Kumar, Hitendra K. Malik, Anima Ghosh, R. Thangavel, K. Asokan,An insight to origin of ferromagnetism in ZnO and N implanted ZnOthin films: Experimental and DFT approach [J],Journal of Alloys and Compounds 768 (2018) 323-328
Romaka, V.V. Romaka, N. Melnychenko, Yu. Stadnyk, L. Bohun, A. Horyn,Experimental and DFT study of the VeCoeSb ternary system[J] ,Journal of Alloys and Compounds 739 (2018) 771-779
Ibáñez, T. Woźniak, F. Dybala, R. Oliva, S. Hernández,R. Kudrawiec,High-pressure Raman scattering inbulk HfS2: comparison of density functional theory methods in layered MS2 compounds (M = Hf, Mo) under compression [J],Scientific reports (2018) 8:12757,DOI: 10.1038/ s41598-018-31051-y
Garwood, N.A. Modine, S. Krishna,Electronic structure modeling of InAs/GaSb superlattices with hybriddensity functional theory [J],Infrared Physics & Technology 81 (2017) 27–31
Eugene S. Kryachko, Eduardo V. Ludena,Density functional theory: Foundations reviewed[J],Physics Reports 544 (2014) 123–239
B. Obot, D.D. Macdonald, Z.M. Gasem,Density functional theory (DFT) as a powerful tool for designing neworganic corrosion inhibitors. Part 1: An overview [J],Corrosion Science 99 (2015) 1–30
科学指南针为超过3000家高校和企业提供一站式科研服务。截止2021年6月:服务1049家高校、2388家企业,提供249所高校研究所免费上门取样服务,平均每天处理样品数5000+、 注册会员数18w+、平均4.5天出结果、客户满意度超过98%。
免责声明:部分文章整合自网络,因内容庞杂无法联系到全部作者,如有侵权,请联系删除,我们会在第一时间予以答复,万分感谢。








