









 您已经拒绝加入团体
您已经拒绝加入团体

 2025-11-13
2025-11-13
 2253
2253
 0
0
【摘要】 广西师范大学团队在《Advanced Functional Materials》发表研究,通过优选晶体取向优化Co₉S₈/MoS₂异质结构,实现钠离子电池超快速存储。科学指南针提供DFT计算支持,助力机制解析与材料设计。
广西师范大学潘齐常、郑锋华教授团队联合三峡大学李涯浩、中南大学欧星教授团队在《Advanced Functional Materials》发表创新研究成果,通过优选晶体取向优化Co₉S₈/MoS₂异质结构,成功实现钠离子电池超快速存储性能。科学指南针为本研究提供密度泛函理论计算支持,助力钠离子存储机制解析与材料设计优化。
研究背景与钠离子电池挑战
钠离子电池(SIBs)因钠资源丰富、成本低廉成为锂离子电池的理想替代者,但在高功率快速充放电场景下面临严峻挑战。钠离子的大离子半径和缓慢电化学反应动力学制约其实际应用性能。
核心技术瓶颈:
-
钠离子半径大导致扩散动力学缓慢
-
电极材料在充放电过程中体积膨胀严重
-
循环稳定性差,容量衰减快
-
高倍率性能不足,难以满足快速充放电需求
创新方法:晶体取向优化策略
研究团队提出通过优选晶体取向优化异质结构的设计策略,成功制备具有各向同性结构的Co₉S₈/MoS₂异质结,突破钠离子存储性能限制。
技术突破要点:
-
设计各向同性晶体结构,提供快速钠离子扩散通道
-
构建Co₉S₈/MoS₂异质结,优化界面电子结构
-
实现内应力均匀分散,增强结构稳定性
-
协同效应提升钠离子传输效率和存储容量
.png)
材料设计与结构表征
通过精确控制合成条件,成功制备具有优选晶体取向的Co₉S₈/MoS₂异质结微米棒结构,并通过多尺度表征手段确认其结构特征。
结构特征验证:
-
HRTEM显示MoS₂的(002)晶面呈现各向同性取向
-
异质界面形成明确晶界,促进电荷传输
-
元素分布均匀,Co₉S₈与MoS₂晶格匹配良好
-
棒状结构在硫化后完好保留,提供稳定框架
.png)
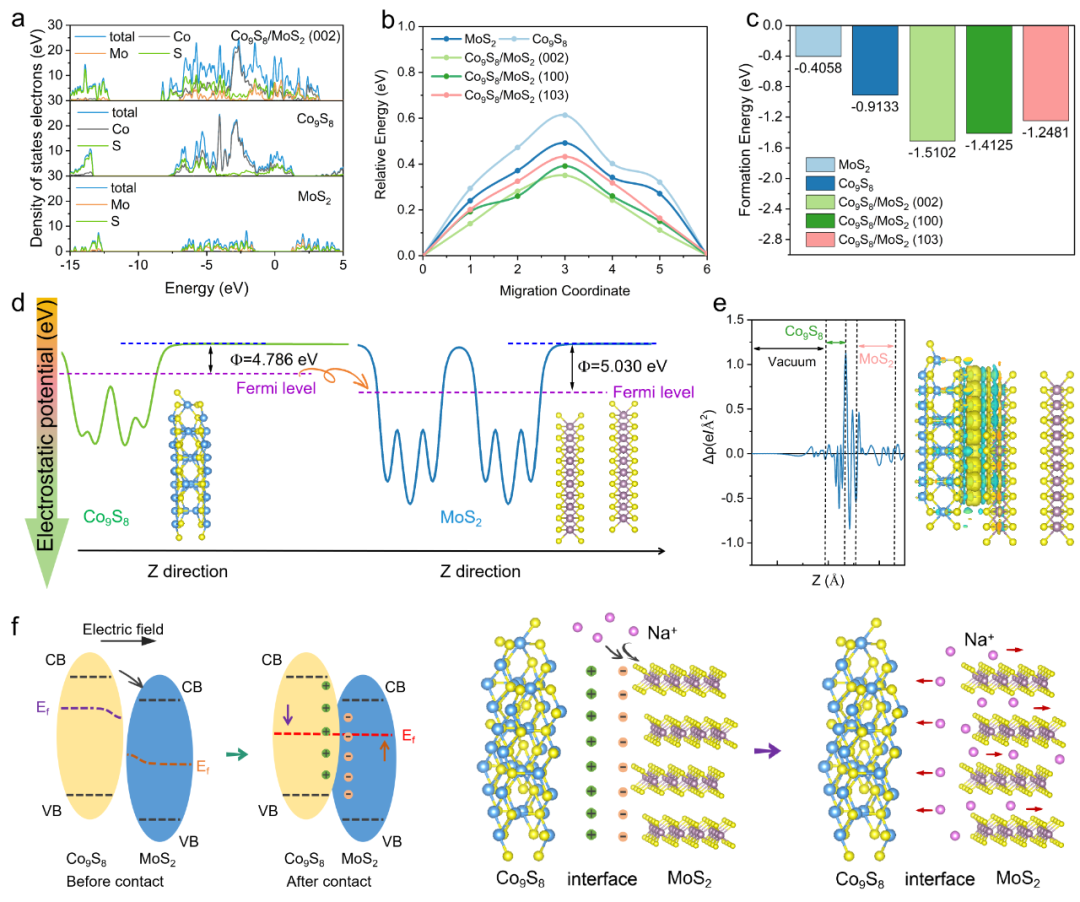
理论计算与机制解析
密度泛函理论计算深入揭示异质结电子结构调控机制和钠离子传输增强原理。
计算研究发现:
-
MoS₂的(002)晶面与Co₉S₈相互作用最强
-
界面电荷转移显著,钠离子迁移能垒降低
-
功函数差异驱动电子自发迁移(Co₉S₈:4.786 eV, MoS₂:5.030 eV)
-
高浓度梯度驱动钠离子双向扩散
-
各向同性结构促进钠离子均匀分布
.png)
电化学性能评估
系统评估Co₉S₈/MoS₂异质结钠离子存储性能,展现出卓越的倍率性能和循环稳定性。
性能卓越表现:
-
2.0 A g⁻¹电流密度下循环2000次容量保持492.1 mAh g⁻¹
-
15.0 A g⁻¹超高倍率下容量达504 mAh g⁻¹
-
首次库仑效率高达92.1%,副反应影响小
-
100次循环后容量保持663.8 mAh g⁻¹
-
赝电容贡献率91%,主导钠离子存储机制
.png)
储能机制与结构演变
通过原位表征技术深入研究钠离子存储机制和材料结构演变规律,揭示优异性能的本质原因。
机制研究突破:
-
原位XRD和拉曼证实氧化还原反应可逆性
-
钠化阶段通过转化和插层反应生成中间产物
-
脱钠过程可逆恢复原始组分,异质界面保持完整
-
电荷转移电阻在循环中逐渐降低,动力学稳定性优异
-
各向同性结构有效缓解钠化过程应力集中
.png)
全电池性能验证
组装NVP@C//Co₉S₈/MoS₂全电池验证实际应用性能,展现出优异的循环稳定性和倍率性能。
应用性能表现:
-
0.5 A g⁻¹循环300次容量保持333 mAh g⁻¹
-
1.0 A g⁻¹循环500次容量维持318 mAh g⁻¹
-
成功点亮LED,证明实际应用潜力
-
倍率性能显著优于已报道钠离子全电池
-
在宽电流范围内保持高可逆容量
.png)
总结与展望
本研究通过优选晶体取向优化异质结构设计策略,为高性能钠离子电池电极材料开发提供创新解决方案。
创新价值总结:
-
各向同性晶体结构提供快速离子传输通道
-
异质结协同效应优化界面电子结构和离子传输
-
内应力均匀分散增强结构稳定性和循环寿命
应用前景:
-
为高功率钠离子电池开发提供新材料体系
-
推动钠离子电池在储能电站和电动汽车中的应用
-
为其他多价离子电池电极材料设计提供借鉴
-
促进低成本、高性能储能技术的发展
论文信息:Advanced Functional Materials, 2025
DOI:10.1002/adfm.202500165
科学指南针计算服务:提供密度泛函理论计算与分子模拟支持,助力储能材料设计与性能优化。了解更多:https://www.shiyanjia.com/simulate.html【科学指南针·服务声明·2025】








