









 您已经拒绝加入团体
您已经拒绝加入团体

 2021-06-24
2021-06-24
 14521
14521
 0
0
【摘要】 回看异质结搭建对不同形状界面处理方法那部分
在做模拟计算时,科学指南针检测平台工作人员在与很多同学沟通中了解到,好多同学对此项目不太了解,针对此,科学指南针检测平台团队组织相关同事对公开课进行问题收集并整理,希望可以帮助到科研圈的伙伴们;
1.请问FCC结构怎么截取111平面并是矩形
回看异质结搭建对不同形状界面处理方法那部分
2.XSD文件名字怎么设置的?
随意设置,别乱改文后缀名就好
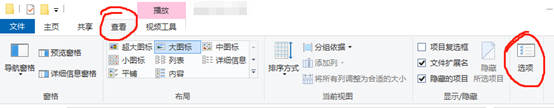
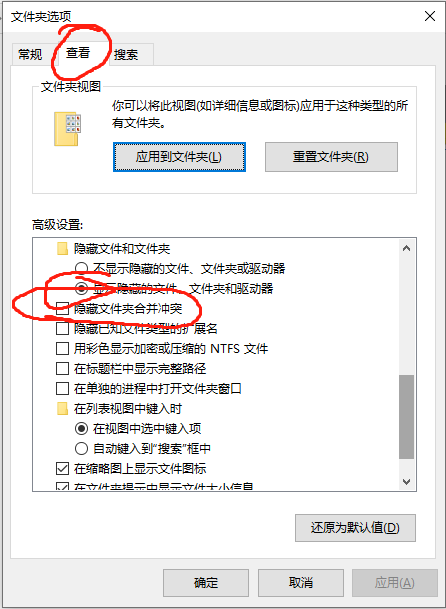
3.请问隐藏的文件那个怎么设置来着
随意打开一个文件夹
4.计算成功了但没有outmol文件怎么办?
如果是可以尝试右击jobs窗口里的任务重新下载试试。或者直接去服务端jobs保存文件夹下去找。
如MS2019 windows服务端在
安装目录\BIOVIA\Materials Studio 19.1 x64 Server\etc\Gateway\root_default\dsd\jobs
MS2019 linux服务端在
安装目录/BIOVIA/MaterialsStudio19.1/etc/Gateway/root_default/dsd/jobs
5.非晶的怎么搭呢
回看第三讲界面吸附模型(指南针学院schooin可看回放)
6.请问怎么搭建合金的超晶胞?
建议先建立合金再进行扩胞,合金的搭建见第三讲。可以使用disorder脚本枚举建立,也可以通过手动替换原子得到
7.请问下配位数怎么求呢?
建议查阅相关文献或者书籍,这个并没有一个肉眼看就能确定的标准
8.左下角的只有project 窗口怎么办
View-Explorers-勾选全部三种Explorer
9.这个结构不合理啊,一个碳上连五根键?
仅是演示建模
另外量子化学,第一性原理等基于量子力学的计算 不用在意几重键的问题,只需要关注是不是多了或者少了原子以及位置是否合理。
10.是根据什么计算化学键数量的呀,能量吗
进行键级分析才可以知道。做计算化学请忘记脑海中关于形式键级那些概念
11.这一步操作的时候出现的问题
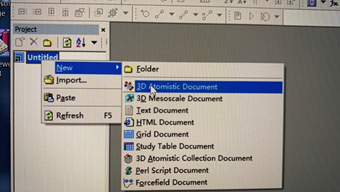
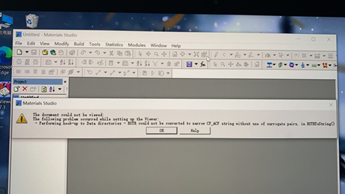
仔细检查是不是将project建立在了含有中文的路径下
12.已知物质结构是否可以用ms计算出量?
看不懂这个问题
13.120°90°90°是怎么确定的?
左下角属性lattice 3D窗口查看得到的
- MOF搭建
MOF搭建,确定晶胞,然后绘制好片段,然后再填充到晶胞中,并仔细修改各种连键,然后优化即可。
15.质子化后的胍基咋画电荷每个N分配1/3?
计算后做原子的布居分析后才知道
16.四氧化三锰团簇怎么画呀请问
个人推荐,对于这种常见物质,直接去数据库下载结构就好。
17.forcite可以优化有机药物吗
选择正确的力场就可以准确优化结构,如果要研究电子相关性质需要用量子力学为基础的相关理论工具,而不能使用Forcite
18.老师,有机聚合物可以去哪个网站下载?
这个需要自己搭建。
19.聚合物放入P1晶胞后,晶格大了怎么处理呢
不清楚这个情况具体发生细节,不过建议回看第三讲界面吸附模型的讲解,尤其注意我对聚合物非晶胞的搭建简易
20.COD 数据库网址可以发一次吗
http://www.crystallography.net/cod/
21.问一下 MS里的建模然后力场优化与chem3d里建模优化有什么区别
Chem3D里面的优化与动力学功能支持的理论方法很少,建议不要做为计算工具
22.chem3D粘贴到MS能演示一下吗
Chem3D保存结构为PDB然后将PDB文件拖入MS的project窗口中就可以
23.金属氧化物cif里氧原子多了怎么办?
分情况来看,如果是分数占据导致的错误,可以看模拟计算联盟公众号关于分数占据体系的讲解。如果单纯就是搭建时候手画导致的,直接删掉就好,删掉时候注意一下,留心其他位置氧原子是否也因为对称性被同时删去
本文所有内容文字、图片和音视频资料,版权均属科学指南针网站所有,任何媒体、网站或个人未经本网协议授权不得以链接、转贴、截图等任何方式转载。








