









 您已经拒绝加入团体
您已经拒绝加入团体

 2024-08-01
2024-08-01
 3004
3004
 0
0
【摘要】 在这项工作中,Zhang等人[1]使用密度泛函理论(DFT)计算和从头算分子动力学模拟,发现在BVO中,VO4四面体结构非常稳定,并且存在很强的表面重构,导致表面上的V原子具有与体中相同的坐标。
钒酸铋(Bismuth vanadate, BiVO4/BVO)由于其易于合成和合适的带隙(2.4 eV)等优点。近年来,作为光催化水分解半导体材料得到了广泛的研究。然而,BVO仍有一些缺点,其中之一是光催化水氧化活性较低。有趣的是,在目前的文献中,Bi原子被作为析氧反应(OER)的活性位点,而V金属原子没有在OER中进行研究,其根本原因尚不清楚。
在这项工作中,Zhang等人[1]使用密度泛函理论(DFT)计算和从头算分子动力学模拟,发现在BVO中,VO4四面体结构非常稳定,并且存在很强的表面重构,导致表面上的V原子具有与体中相同的坐标。对于一些高折射率表面,理论上预测存在一些不饱和V位,但从水中取氧原子,很容易重新形成VO4四面体结构。OER的其他中间体很难在这种VO4结构上吸附或解吸,这使得BVO中的V位点不适合作为OER的活性位点。
对于所有曲面,在几何优化中使用基元单元,并设置相应的 K 点。超胞表面用于OER计算。六个 Bi 层用于松弛层,如 (001) 表面。我们还使用了八层进行测试,发现能量差仅小于0.01 eV。对于其他曲面,使用与构建 (001) 曲面的策略类似的策略。首先,松弛层的长度几乎与(001)表面的长度相同。然后,再使用两层进行测试,直到能量差小于0.01 eV。如果表面的顶层和底层相同,则该表面是对称的表面,并且不添加偶极校正。如果表面的顶层和底层不同,添加偶极校正。
对于OER计算,使用Nørskov的四步PCET协议,中间体表示*、OH*、O*和OOH*,OER过电位由下式确定:
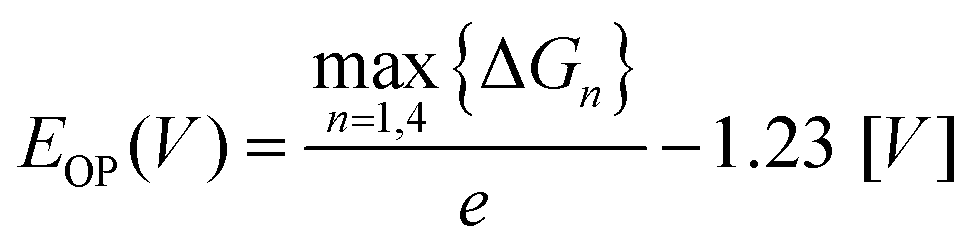
在300和673 K的分子动力学模拟中,该VO4结构保持稳定。图1中表示所有的表面都是由BVO的单元格构建的。以该重构表面为模型,在300 K下进行分子动力学。仿真结果如图2所示。在300k时,模拟过程中的能量变化很小。顶部表面的VO4结构保持其四面体形状,并始终作为一个整体运动。各种BVO形态的XPS表征验证了该研究从DFT和分子动力学模拟中得到的主要发现。
结果表明,BVO表面存在不饱和的Bi位点,而不饱和的V位点未被观察到。该研究为BVO的OER活性的增强提供了新的见解,并为其他含V原子光催化剂的OER活性提供了基本的理解。
BVO的单元格,以及常用的(b)(001)和(c)(101)表面。.png)
图1(a)BVO的单元格,以及常用的(b)(001)和(c)(101)表面。大红色、小红色和紫色球体分别是V、O和Bi原子
表面的能量变化.png)
图2 300 K分子动力学模拟时BVO(101)表面的能量变化。
[1]. Zhang, Q.; Liu, G.; Liu, T., Oxygen evolution reaction (OER) active sites in BiVO4 studied using density functional theory and XPS experiments. Physical Chemistry Chemical Physics 2024, 26 (3), 2580-2588.
科学指南针充分发挥互联网技术和业务优势,在国内率先打造出业界领先的线上化、数字化的科研服务基础设施,在行业内首创用户自主下单、服务全流程追踪、测试“云现场”等模式,进一步提高了大型科学仪器设施开放共享和使用效率,以实际行动助力科技创新。现已发展成为中国专业科研服务引领者,已获得检验检测机构资质认定证书(CMA)、实验动物使用许可证、“ISO三体系认证”等专业认证。
免责声明:部分文章整合自网络,因内容庞杂无法联系到全部作者,如有侵权,请联系删除,我们会在第一时间予以答复,万分感谢。








