









 您已经拒绝加入团体
您已经拒绝加入团体

 2021-07-29
2021-07-29
 7624
7624
 0
0
【摘要】 本文仅提供一个我们日常工作中的处理思路,不能面面俱到,适用场景请大家自行变通。 本文不涉及到晶体解析或者模拟计算上的枯燥概念,一切仅以说清楚事情为标准去叙述。
本文仅提供一个我们日常工作中的处理思路,不能面面俱到,适用场景请大家自行变通。
本文不涉及到晶体解析或者模拟计算上的枯燥概念,一切仅以说清楚事情为标准去叙述。
为什么会有分数占据的情况存在?
晶体解析出来的结构必然是和自然界中的晶体实际结构有关的,而不会去考虑你之后是否用这个cif文件去画图或者计算而“自我优化”或者“人为优化”。大家拿到晶体解析得到的带分数占据的cif不要惊讶,那才是真实的晶体的状态。
大家都知道,常规的单晶解析中,H原子都是“猜”上去的,因为测不准H原子位置,那么只能把重原子(非H的都算)位置确定后,然后根据化学环境去猜,所以大家拿到的cif里面的可能会出现any-H键过长或者过短的情况,都是猜H的软件的问题。所以在进行计算前都要经过几何优化或者限制性优化(冻结非H后优化)。
如果某处有个甲基,我们知道甲基的C-C键会自由旋转,那么此处的H应该是不确定的,理应出现分数占据那种无序的状态,真的要弄起来可能你看到的是甲基旋转时H的慢动作投影图。但是因为前面说了H是猜上去的,大家知道此处是自由旋转的时候也只会猜三个整数占据的H上去。
那如果是叔丁基会怎么样?此时原本随性惯了的H变成了实打实的甲基。如果此时没有其他周围空间的限制,与环相连的C-C键会因为旋转,导致末端的可能性增多,至少2种主要构象存在。
其实最常见的是晶格内溶剂分子无序。受到大环境约束,溶剂分子不可能多出一个,但是活动空间还是有一些的。如果直接用VESTA打开那种晶体cif,你会看到溶剂分子部位非常鬼畜。
.png)
还有一种情况,比如培养的过程即使用了CHCl3和 CH2Cl2两种溶剂,然后解析时候发现无论以哪个去对应溶剂分子都有问题,那个时候可能就会解出分数占据。这时候不是分子无序导致的分数占据,而是两种溶剂在同一个位置有体现,反应出的统计量上面的效果就是那种说谁都不是的情况。这不是错,这是对真实情况的反应。当然也会出现溶剂格子内溶剂分子丢失的情况,比如部分脱水(溶剂)。
怎么处理分数占据使其能够用于计算
一般我的处理思路是“保大去小”。
首先可以用VESTA打开无序的cif(这里的cif是单晶解析已经结束并且无错的cif,只是计算人员不满意的无序状态)。然后另存为cif。
这里推荐使用VESTA是因为,该软件会对分数占据原子位置使用饼状图表示,很容易辨别高占据低占据,更同意梳理思路。这么做是为了清洗一下cif中的对我们计算来说无效的数据。原始的cif中数据很多,但是会严重干扰我们修改cif。
然后用记事本工具打开cif
找到occupancy定义位置。这里我们可以看到_atom_site_occupancy在第二行,那么下面数据区的第二列就是占据数。可以看出这里的占据是0.176 0.25……这些占据数。
.png)
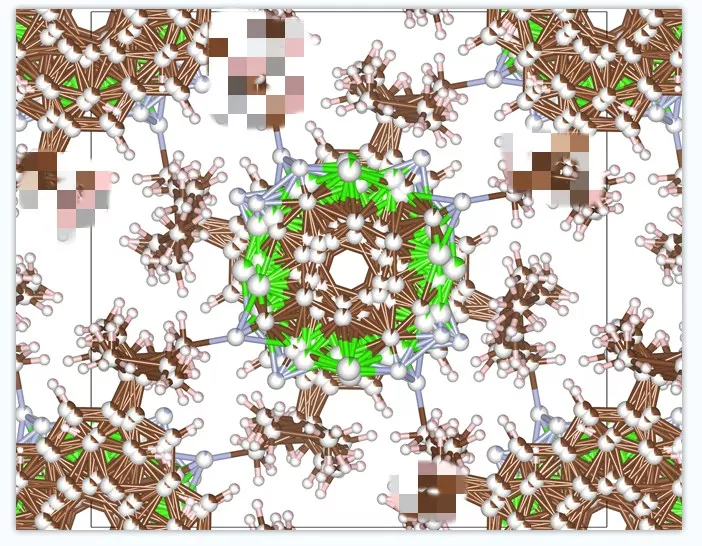
如果此时出现的是0.8和0.2 ,可以直接删除所有0.2占据的原子,然后保存并用VESTA打开,发现其中没有原子丢失就可以放心吧0.8数值都改成1.0,然后再保存。就得到了一个对于晶体解析不算合理但是对于计算模拟和自己画图很适合的cif文件。
当然VESTA提供了更方便的功能
.png)
可以直接删除掉低占据的原子,然后把高占据的改为1就好了。
以上的演示都不属于这种简单情况(因为简单修改的cif我这边没有保存,只能拿复杂体系截图做教程了)
如果你手头的cif不属于简单情况,比如有好几组分子是0.25占据,出线4选1情况,此时无法通过占据数做简单归类,那么需要认真手动修改了。
首先要清楚知道自己的cif中到底是有什么物质,然后就是把那些混在一起的物质抽出一个 然后改为1占据就好了。
将无序cif导入MS,然后显示成键(以一个不乱的阈值去显示)。
.webp)
然后直接删除多余的就好了。
这里有三个建议。
1.不要直接删除多余的结构,而是先用line模式显示所有的原子。然后挑选可能的原子并用ball and stick显示。一堆找完了,此时再放心删去多余的。
2.在你修改过程发现有些结构被cell边缘的一些结构遮挡了,可以通过display style-lattice里面调整ABC的range来暂时隐藏掉那些边缘结构。
.png)
3.时时刻刻注意属性窗口中每个原子的占据数,不同的占据数的原子不可能是在同一个分子片段中的,必须要注意,以免前功尽弃。
.webp)
科学指南针这边的单晶解析出来的cif是具有正确的空间群约束的,所以删除的过程中会很快。因为对称的原因,一般你只需要修改看到的无序的结构不到3成的原子就能搞完了。
那有没有主动构造分数占据的情况?
有,比如研究合金的时候。1%掺杂的合金,那么必须要构建一个含有100原子的格子才能体现出这种1%掺杂。如果是使用了虚拟晶体近似 (Virtural Crystal Approximation,VCA) 的方法,则模型里面的原子就可以指定成如0.1A元素加0.9B元素这种样子,可以以最小晶胞来做计算用的模型。目前CASTEP支持该方法,但是该方法有很多局限性,如果大家感兴趣就点一下赞,我们以后就说说怎么做合金计算和应该要注意的一些事项。
本文所有内容文字、图片和音视频资料,版权均属科学指南针网站所有,任何媒体、网站或个人未经本网协议授权不得以链接、转贴、截图等任何方式转载。








