









 您已经拒绝加入团体
您已经拒绝加入团体





 可享受最低
可享受最低 元的信用额度,先测后付0元下单
元的信用额度,先测后付0元下单

 元无门槛现金免单券
元无门槛现金免单券
 积分加速得,千万好礼等你兑
积分加速得,千万好礼等你兑














第一性原理
87.1%
好评率
部分论文致谢
项目简介
第一性原理计算的基本思想是将多个原子构成的体系看成是由多个电子和原子核组成的系统,并根据量子力学的基本原理对问题进行最大限度的“非经验性”处理。它只需要5个基本常数(m0,e,h,c,kB)就可以计算出体系的能量和电子结构等物理性质。它可以确定已知材料的结构和基础性质,并实现原子级别的精准控制,是现阶段解决实验理论问题和预测新材料结构性能的有力工具。并且,第一性原理计算不需要开展真实的实验,极大地节省了实验成本,现已被广泛应用于化学、物理、生命科学和材料学等领域。
适合的研究方向包括但不限于:催化、电池、半导体、金属材料、非金属材料、合金、纳米材料等
可以计算的体系包括但不限于:晶体、非晶、二维材料、表面、界面、固体等
常用软件:VASP,MS,CP2K,QE等
可以计算的内容包括但不限于:
材料的几何结构参数(如键长、键角、二面角、晶格常数、原子位置等)
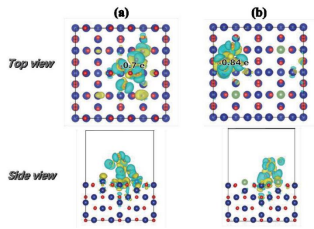
材料的电子结构信息(如电荷密度、电荷差分密度、态密度、能带、费米能级、功函数、ELF等)
材料的光学性质(如介电常数等)
材料的力学性质(如弹性模量等)
材料的磁学性质(如磁导率等)
材料的晶格动力学性质(如声子谱等)
材料的表面性质(如吸附能,催化计算等)
复合材料的性质(异质结等内容)等等
{{moduleItem.modulename}}
样品要求
最好提供晶体结构文件,即cif文件;
或者可以提供与XRD实验数据匹配的PDF卡片信息;
如果是表面结构,还需要提供晶面信息;如结构有改性(空位、掺杂、位错等),还需要说明细节
常见问题

1. 为什么能带计算值一般低于实验值?
这是PBE本身的问题,PBE是一种GGA泛函,有较强的自相互作用问题,导致电子偏向离域、带隙低估等一系列问题。一般计算得到的带隙比实验低30%~50%。这种低估并不是说方法就不可以用,我们还是可以把这个方法拿来做定性分析。
解决办法,方法有DFT+U、mBJ+U、杂化泛函
2. 为什么有些体系要加U?










