









 您已经拒绝加入团体
您已经拒绝加入团体

 2025-04-10
2025-04-10
 4534
4534
 0
0
【摘要】 本文通过密度泛函理论分析Li+配体结合能变化,揭示氟化对锂电池电解质性能的影响机制,为高功率电池研发提供理论支持。含配位构型可视化及能量数据对比。
在锂离子电池技术迭代过程中,电解质材料的改良始终是提升电池功率密度的关键突破点。本研究通过密度泛函理论(DFT)计算,系统解析了乙腈(ACN)与四氢呋喃(THF)及其全氟类似物与Li+的配位特性,为新型氟化电解质的开发提供重要理论支撑。
一、实验方法与计算模型
采用B3LYP混合泛函进行理论计算,基于6-31+G基组完成主要运算,并通过相关一致极化价三重zeta基组验证结果精度。所有分子构型经高斯09软件优化后,使用Jmol进行三维结构可视化(图1)。
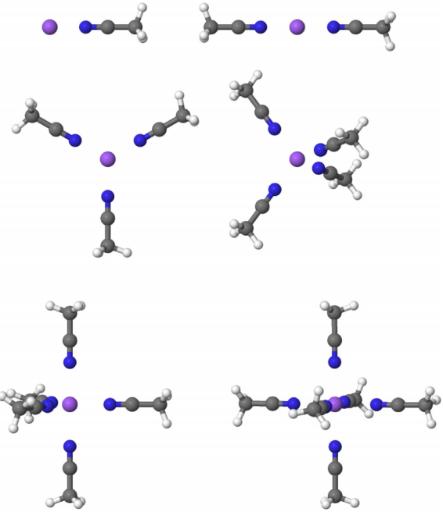
图1 优化结构
二、配位构型与能量特征
1.乙腈体系配位规律
- 单配位体系呈现C3v对称性
- 四配位时达到Td对称
- 六配位体系中重原子呈现近似Oh对称
2.氟化改性影响机制
- 全氟化使偶极矩显著降低(ACN:4.00D→1.18D;THF:2.01D→0.29D)
- Li-N键距增加0.02-0.05Å(表2数据)
- 单配位结合能降低12.1kJ/mol(ACN体系)
n%E5%8F%8A%E5%85%B6%E5%85%A8%E6%B0%9F%E7%B1%BB%E4%BC%BC%E7%89%A9%E7%9A%84%E4%BC%98%E5%8C%96%E7%BB%93%E6%9E%84.png)
图2Li+ (THF)n及其全氟类似物的优化结构
三、氟化改性的双刃剑效应
1.电子效应主导
- 氟原子强吸电子性降低配位原子电荷密度
- 电荷诱导偶极贡献削弱配位强度
2.空间位阻补偿
- 多配位体系中配体间斥力降低(五配位能量差缩减至14.2kJ/mol)
- 氟化改性的立体效应优化配位构型
四、锂电池电解质设计启示
1.四配位体系展现最佳能量-空间平衡
2.氟化改性需控制取代度以平衡溶解性与离子电导率
3.新型电解质开发应关注:电荷分布重构、配位层稳定性、溶剂化能调控
参考文献:[1] Charles W. Bauschlicher, Li+-ligand binding energies and the effect of ligand fluorination on the binding energies, Chemical Physics Letters, Volume 694, 2018, Pages 86-92.
科学指南针在全国建立32个办事处和20个自营实验室,拥有价值超2.5亿元的高端仪器。检测项目达4000+项,覆盖材料测试、环境检测、生物服务、行业解决方案、模拟计算等九大业务。累计服务1800+个高校、科研院所及6000+家企业,获得了60万科研工作者的信赖。
免责声明:部分文章整合自网络,因内容庞杂无法联系到全部作者,如有侵权,请联系删除,我们会在第一时间予以答复,万分感谢。








