









 您已经拒绝加入团体
您已经拒绝加入团体

 2025-06-09
2025-06-09
 3194
3194
 0
0
【摘要】 解析OER/ORR催化剂中OH*/OOH*吸附自由能标度关系,探讨DFT计算系统误差的修正方法,揭示最小过电位与活性预测规律,为燃料电池/电解槽催化剂设计提供理论依据。
开发高效、稳定的析氧反应(OER)和氧还原反应(ORR)催化剂,是推进水电解槽和燃料电池商业化的核心挑战。理解催化反应机理需基于精准的理论基础,其中密度泛函理论(DFT)研究揭示了关键规律。
核心发现:OH*/OOH*吸附能标度关系
DFT研究表明,OH与OOH中间体的吸附自由能存在稳定标度关系:
(基于RPBE泛函计算)。与实验值(3.4 eV)相比,该关系为催化剂设计提供理论依据:
- ORR/OER最佳自由能差:2.46 eV
- 最小热力学过电位:
- ORR最优吸附能:(略低于铂)
标度关系的可靠性与误差分析
±0.2 eV偏差反映不同材料表面的分散性(非计算误差)。但在特定材料(如铂合金)中,分散性显著降低。
关键挑战:DFT对OOH*中氧-氧键的描述存在系统误差(类似过氧化物键难模拟)。此误差可能影响标度关系预测的准确性。
误差修正与改进
通过量化气态过氧化物的系统误差,研究发现:
1.未包含的vdW相互作用与误差部分抵消;
2.修正后标度关系偏移仅差0.1 eV,验证原关系的可靠性;
3.误差修正方法已应用于CO₂还原反应,提升计算精度。
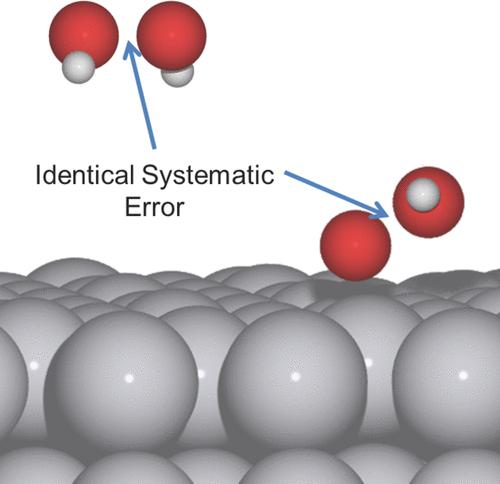
图1 催化机理示意图
结论与展望
标度关系仍是预测催化剂活性的有效工具。通过修正系统误差,可进一步提升DFT在OER/ORR催化剂设计中的指导价值。未来需结合机器学习与高精度计算,优化材料筛选流程。
参考文献:1.Rune Christensen, Heine A. Hansen, Colin F. Dickens, Jens K. Nørskov, and Tejs Vegge, The Journal of Physical Chemistry C 2016 120 (43), 24910-24916, DOI: 10.1021/acs.jpcc.6b09141.
科学指南针以分析测试为核心,提供材料测试、环境检测、生物服务、模拟计算、科研绘图等多项科研产品,累计服务1800+个高校、科研院所及6000+家企业,获得了60万科研工作者的信赖。始终秉持“全心全意服务科研,助力全球科技创新”的使命,致力于为高校、院所、医院、研发型企业等科研工作者提供专业、快捷、全方位的服务。
免责声明:部分文章整合自网络,因内容庞杂无法联系到全部作者,如有侵权,请联系删除,我们会在第一时间予以答复,万分感谢。








